

Symmetric adenine methylation is an essential DNA modification in the early-diverging fungus Rhizopus microsporus
- PMID: 40268918
- PMCID: PMC12019607
- DOI: 10.1038/s41467-025-59170-x
Symmetric adenine methylation is an essential DNA modification in the early-diverging fungus Rhizopus microsporus
Abstract
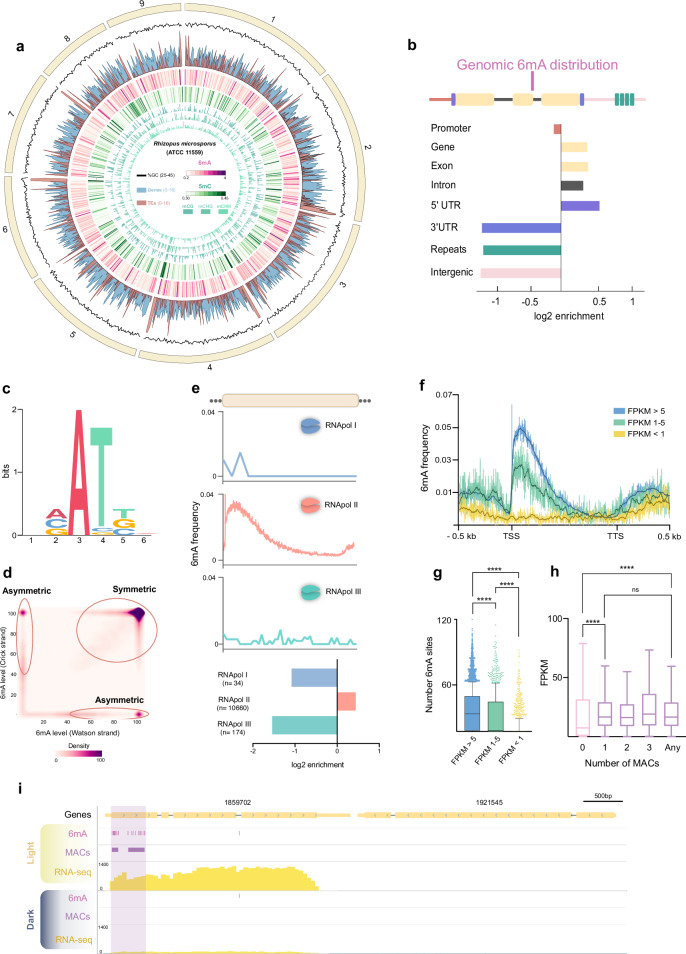
The discovery of N6-methyladenine (6mA) in eukaryotic genomes, typically found in prokaryotic DNA, has revolutionized epigenetics. Here, we show that symmetric 6mA is essential in the early diverging fungus Rhizopus microsporus, as the absence of the MT-A70 complex (MTA1c) responsible for this modification results in a lethal phenotype. 6mA is present in 70% of the genes, correlating with the presence of H3K4me3 and H2A.Z in open euchromatic regions. This modification is found predominantly in nucleosome linker regions, influencing the nucleosome positioning around the transcription start sites of highly expressed genes. Controlled downregulation of MTA1c reduces symmetric 6mA sites affecting nucleosome positioning and histone modifications, leading to altered gene expression, which is likely the cause of the severe phenotypic changes observed. Our study highlights the indispensable role of the DNA 6mA in a multicellular organism and delineates the mechanisms through which this epigenetic mark regulates gene expression in a eukaryotic genome.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures






References
-
- Duncan, E. J., Gluckman, P. D. & Dearden, P. K. Epigenetics, plasticity, and evolution: how do we link epigenetic change to phenotype? J. Exp. Zoöl. Part B Mol. Dev. Evol.322, 208–220 (2014). - PubMed
-
- Adhikari, S. & Curtis, P. D. DNA methyltransferases and epigenetic regulation in bacteria. FEMS Microbiol. Rev.40, 575–591 (2016). - PubMed
