

Genomic and genetic insights into Mendel's pea genes
- PMID: 40269167
- PMCID: PMC12221995
- DOI: 10.1038/s41586-025-08891-6
Genomic and genetic insights into Mendel's pea genes
Abstract
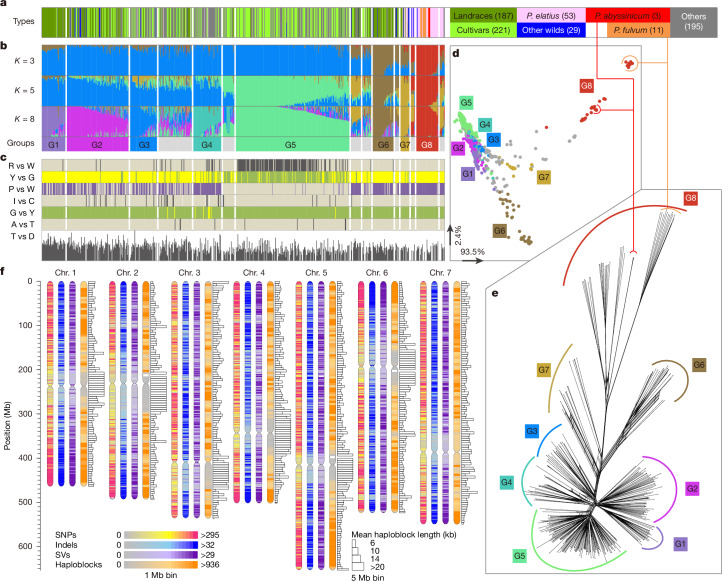
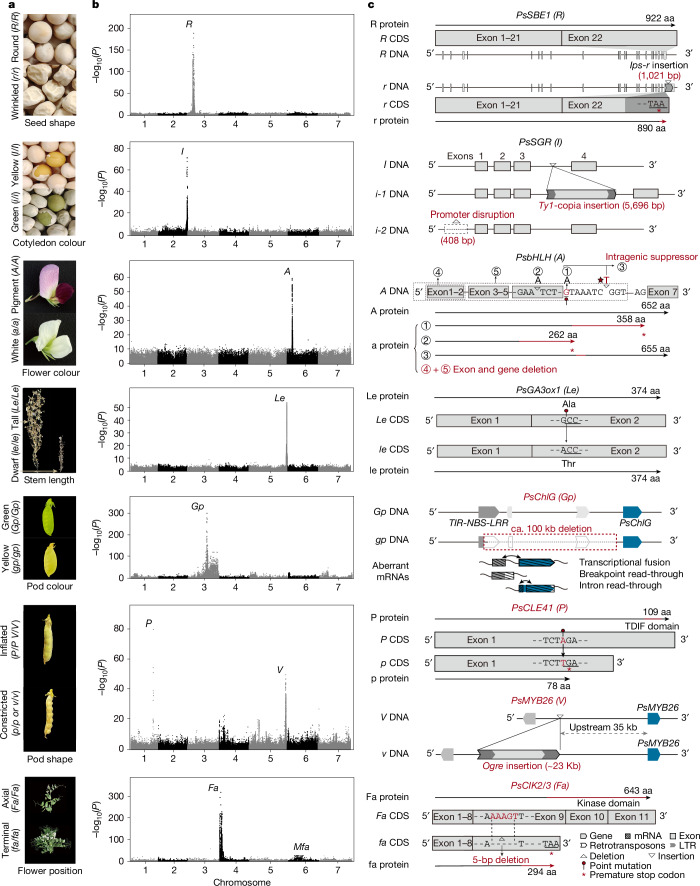
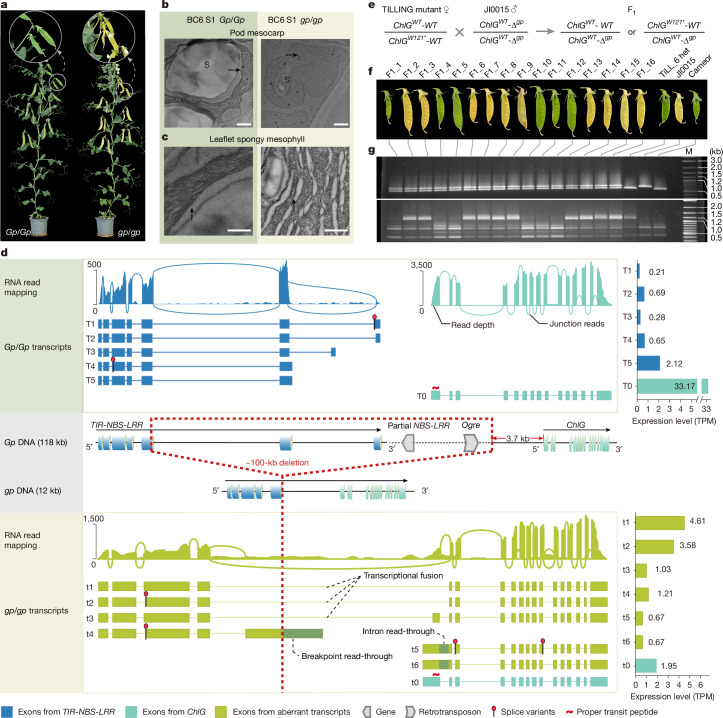
Mendel1 studied in detail seven pairs of contrasting traits in pea (Pisum sativum), establishing the foundational principles of genetic inheritance. Here we investigate the genetic architecture that underlies these traits and uncover previously undescribed alleles for the four characterized Mendelian genes2-7, including a rare revertant of Mendel's white-flowered a allele. Primarily, we focus on the three remaining uncharacterized traits and find that (1) an approximately 100-kb genomic deletion upstream of the Chlorophyll synthase (ChlG) gene disrupts chlorophyll biosynthesis through the generation of intergenic transcriptional fusion products, conferring the yellow pod phenotype of gp mutants; (2) a MYB gene with an upstream Ogre element insertion and a CLE peptide-encoding gene with an in-frame premature stop codon explain the v and p alleles, which disrupt secondary cell wall thickening and lignification, resulting in the parchmentless, edible-pod phenotype; and (3) a 5-bp exonic deletion in a CIK-like co-receptor kinase gene, in combination with a genetic modifier locus, is associated with the fasciated stem (fa) phenotype. Furthermore, we characterize genes and alleles associated with diverse agronomic traits, such as axil ring anthocyanin pigmentation, seed size and the 'semi-leafless' form. This study establishes a foundation for fundamental research, education in biology and genetics, and pea breeding practices.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures









References
-
- Mendel, G. Versuche über plflanzenhybriden. Verh. Naturforsch. Vereines Brünn4, 3–47 (1866).
-
- Bhattacharyya, M. K., Smith, A. M., Ellis, T. H. N., Hedley, C. & Martin, C. The wrinkled-seed character of pea described by Mendel is caused by a transposon-like insertion in a gene encoding starch-branching enzyme. Cell60, 115–122 (1990). - PubMed
-
- Armstead, I. et al. Cross-species identification of Mendel’s i locus. Science315, 73 (2007). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
