

Genomic insights into the population structure, antimicrobial resistance, and virulence of Brachyspira hyodysenteriae from diverse geographical regions
- PMID: 40272172
- PMCID: PMC12131820
- DOI: 10.1128/spectrum.03386-24
Genomic insights into the population structure, antimicrobial resistance, and virulence of Brachyspira hyodysenteriae from diverse geographical regions
Abstract
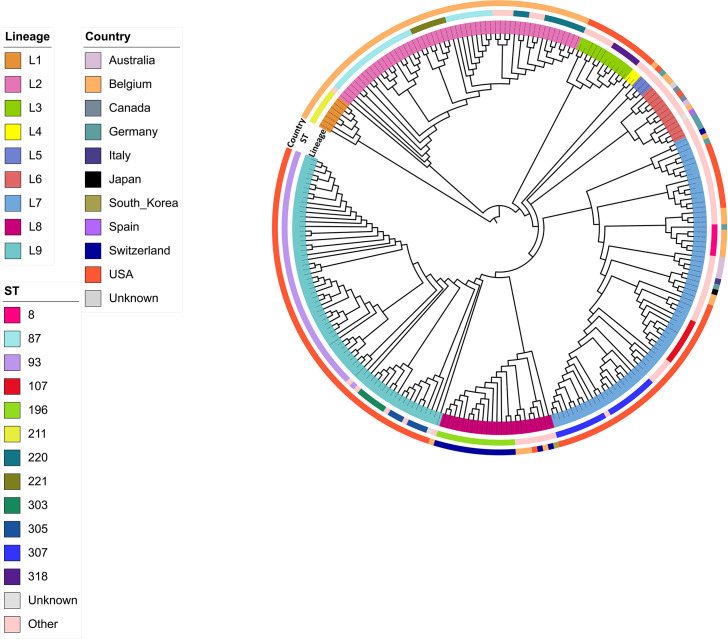
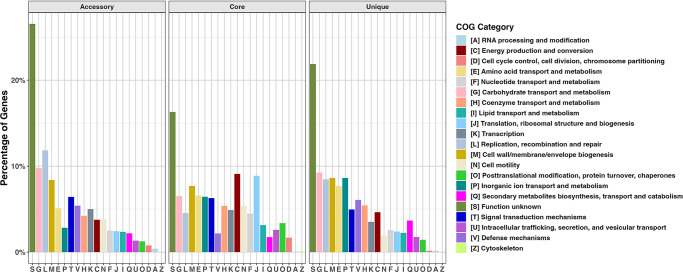
Swine dysentery, caused by the anaerobic spirochete Brachyspira hyodysenteriae, leads to mucohemorrhagic diarrhea in grower-finisher pigs, impacting swine production. Knowledge regarding its genomic epidemiology is limited. We performed a whole-genome sequence analysis for 251 B. hyodysenteriae genomes from 10 countries, including 117 isolates sequenced in this study. Phylogenomic analysis based on core-genome single nucleotide polymorphisms (SNPs) revealed nine lineages, with L7 (72 isolates, 28.69%), L9 (67 isolates, 26.69%), and L2 (53 isolates, 21.12%) predominating. Geographical clustering was observed with distinct lineage distributions. Multilocus sequence typing identified 69 sequence types (STs), including 20 novel STs across 251 genomes. Association between specific lineages, STs, and geographical regions was evident, highlighting evolutionary and regional patterns. The pan-genome analysis identified 5,231 genes, categorized into core (1,648), accessory (2,619), and unique (964) components. Functional annotation linked core genes to essential cellular processes, while accessory and unique genes were enriched in genetic variability, defense mechanisms, and secondary metabolism. The pan-genome exhibited a high proportion of hypothetical genes, necessitating further functional characterization. Antimicrobial resistance (AMR) screening detected the tva(A) and lnu(C) genes associated with tiamulin and lincomycin resistance, respectively, in specific lineages and STs. Virulence factor analysis identified genes linked to hemolysin production, iron uptake, and survival in host environments in most isolates, with a subset of genes demonstrating lineage-specific associations that are further linked to pathogenic potential. This comprehensive genomic epidemiological analysis elucidates the genetic diversity, antimicrobial resistance, and virulence of B. hyodysenteriae globally, enhancing understanding of its epidemiology and guiding interventions to mitigate swine dysentery.
Importance: Brachyspira hyodysenteriae, the primary causative agent of swine dysentery, remains a less-studied pathogen than other bacterial species that impact animal health. This study uses whole-genome sequencing and advanced phylogenomic approaches to reveal the genetic diversity and geographical distribution of B. hyodysenteriae isolates, focusing on U.S. populations. The identification of nine distinct phylogenetic lineages and associated sublineages highlights the pathogen's complex population structure and regional variation. Importantly, the study detects AMR genes, including tva(A) and lnu(C), linked to tiamulin and lincomycin resistance, that may pose significant challenges to disease management. The analysis also identifies virulence-associated genes, shedding light on molecular mechanisms underlying pathogenicity. By combining core-genome SNP phylogenies with multilocus sequence typing and accessory genome insights, this work provides a robust framework for a better understanding of B. hyodysenteriae evolution. Overall, these findings underscore the importance of genomic surveillance in informing control strategies and improving swine health worldwide.
Keywords: AMR; Brachyspira hyodysenteriae; MLST; phylogenomic analysis; population structure; swine dysentery; virulence; whole genome sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
Phylogenetic diversity, antimicrobial susceptibility and virulence gene profiles of Brachyspira hyodysenteriae isolates from pigs in Germany.PLoS One. 2018 Jan 11;13(1):e0190928. doi: 10.1371/journal.pone.0190928. eCollection 2018. PLoS One. 2018. PMID: 29324785 Free PMC article.
-
Understanding the molecular epidemiology and global relationships of Brachyspira hyodysenteriae from swine herds in the United States: a multi-locus sequence typing approach.PLoS One. 2014 Sep 5;9(9):e107176. doi: 10.1371/journal.pone.0107176. eCollection 2014. PLoS One. 2014. PMID: 25192199 Free PMC article.
-
Antimicrobial susceptibility of U.S. porcine Brachyspira isolates and genetic diversity of B. hyodysenteriae by multilocus sequence typing.J Vet Diagn Invest. 2024 Jan;36(1):62-69. doi: 10.1177/10406387231212189. Epub 2023 Nov 15. J Vet Diagn Invest. 2024. PMID: 37968893 Free PMC article.
-
Swine dysentery: aetiology, pathogenicity, determinants of transmission and the fight against the disease.Int J Environ Res Public Health. 2013 May 10;10(5):1927-47. doi: 10.3390/ijerph10051927. Int J Environ Res Public Health. 2013. PMID: 23665849 Free PMC article. Review.
-
A review of methods used for studying the molecular epidemiology of Brachyspira hyodysenteriae.Vet Microbiol. 2017 Aug;207:181-194. doi: 10.1016/j.vetmic.2017.06.011. Epub 2017 Jun 19. Vet Microbiol. 2017. PMID: 28757022 Review.
References
-
- Hampson DJ, Burrough ER. 2019. Swine dysentery and Brachyspiral colitis, p 951–970. In Diseases of swine, 11thWiley Blackwell ed. Wiley Blackwell.
-
- Rugna G, Bonilauri P, Carra E, Bergamini F, Luppi A, Gherpelli Y, Magistrali CF, Nigrelli A, Alborali GL, Martelli P, La T, Hampson DJ, Merialdi G. 2015. Sequence types and pleuromutilin susceptibility of Brachyspira hyodysenteriae isolates from Italian pigs with swine dysentery: 2003-2012. Vet J 203:115–119. doi:10.1016/j.tvjl.2014.10.033 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
