

Using chanarin-dorfman syndrome patient fibroblasts to explore disease mechanisms and new treatment avenues
- PMID: 40275410
- PMCID: PMC12020101
- DOI: 10.1186/s13023-025-03711-6
Using chanarin-dorfman syndrome patient fibroblasts to explore disease mechanisms and new treatment avenues
Abstract
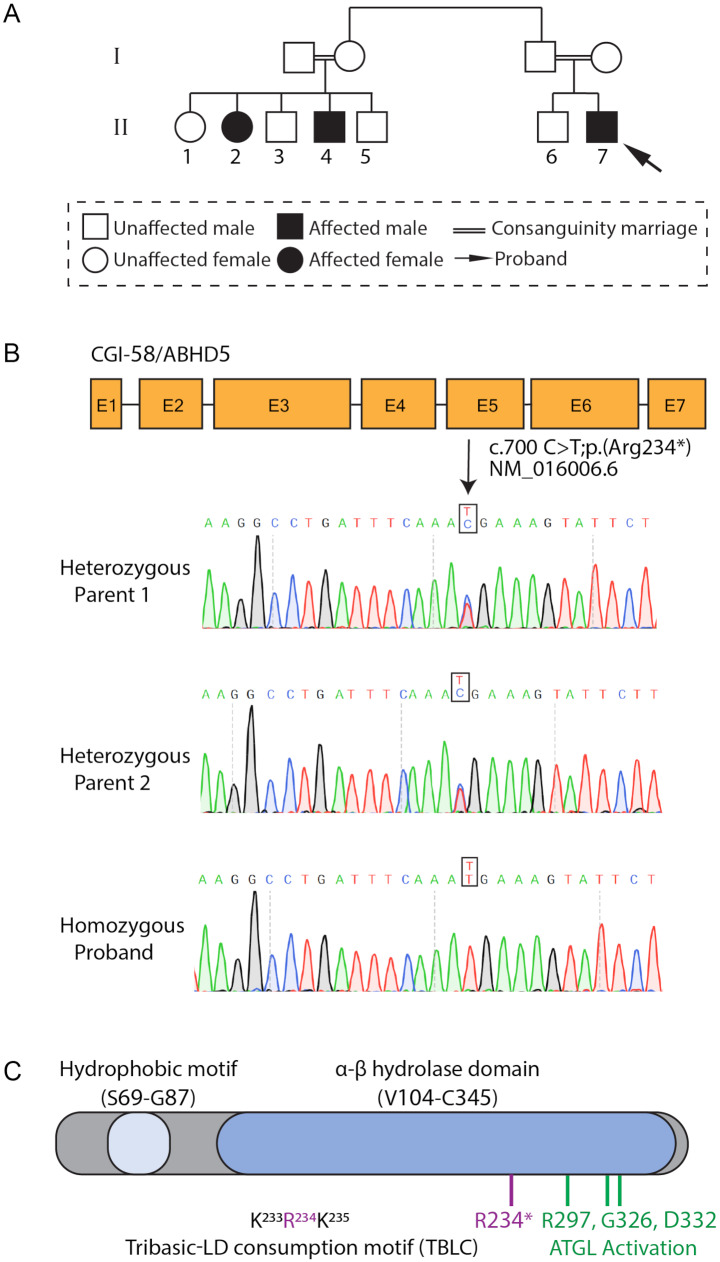
Background: Chanarin-Dorfman syndrome (CDS) is a multisystemic autosomal recessive rare disorder. CDS is caused by variants in the abhydrolase domain containing 5 (ABHD5) encoding gene (CGI-58), which ultimately leads to excessive lipid storage, and therefore a high abundance of cellular lipid droplets (LDs). Although the molecular etiology of the disease was described many years ago, no treatment for CDS is currently available.
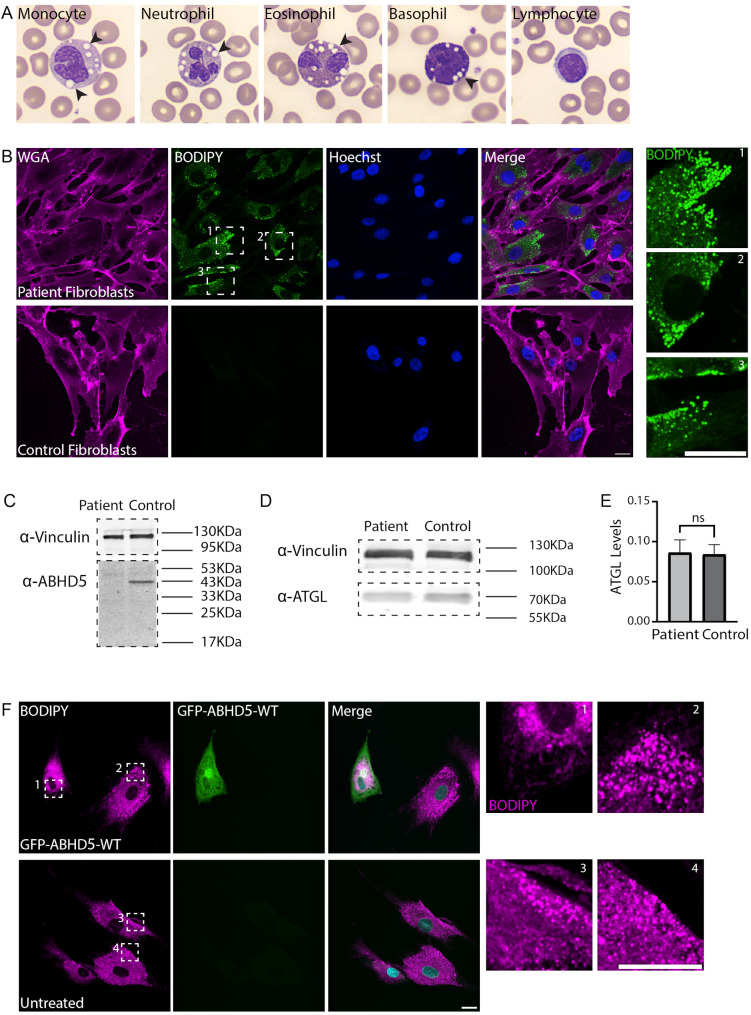
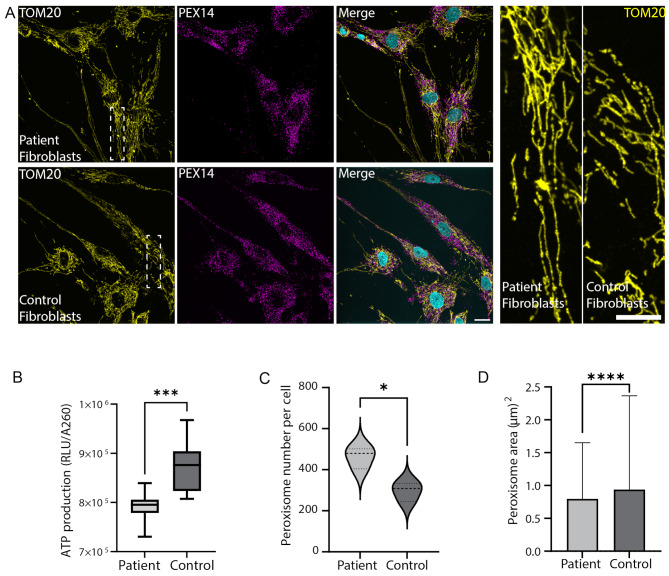
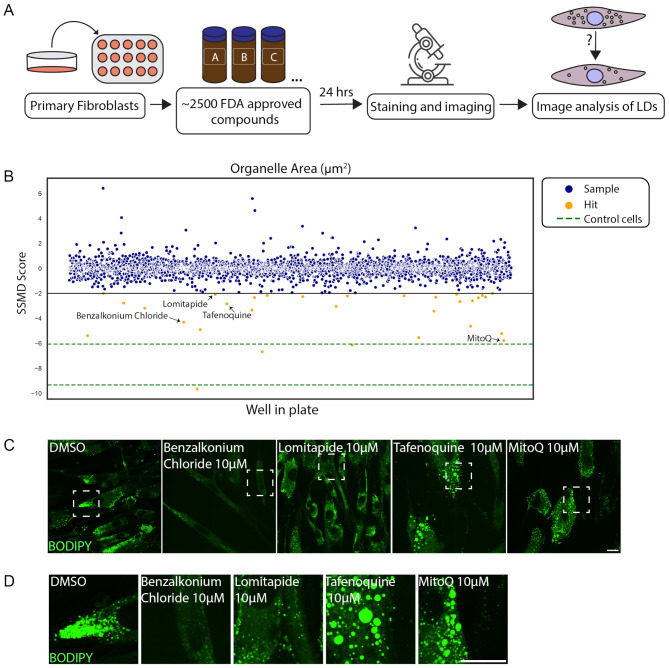
Results: To further characterize the molecular basis of the disease and to uncover new treatment avenues, we used skin fibroblasts originating from a young patient diagnosed with CDS due to a homozygous nonsense mutation. We show that dysfunctional ABHD5 does not only affect LDs, but also influences other metabolic-related organelles; the mitochondria and peroxisomes. Additionally, we found that expressing functional ABHD5 in CDS patient cells reduced LD number. Finally, we developed and applied a high content-based drug repurposing screen based on a collection of ∼2500 FDA approved compounds, yielding several compounds that affected LD total area and size.
Conclusions: Our findings enhance the understanding of the dysfunction underlying CDS and propose new avenues for the treatment of CDS patients.
Keywords: Chanarin-Dorfman syndrome; Drug repurposing; Lipid droplets; Mitochondria; Neutral lipid storage; Peroxisomes.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Family consented to this study according to an approved institutional review board (IRB) protocol (0038 − 14). Consent for publication: The patients’ parents signed on a consent for publication. Competing interests: The authors have no conflict of interest.
Figures
References
-
- Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab. 2009;297(2):E289–96. - PubMed
-
- Nur BG, Gencpinar P, Yuzbasioglu A, Emre SD, Mihci E. Chanarin-Dorfman syndrome: Genotype-Phenotype correlation. Eur J Med Genet. 2015;58(4):238–42. - PubMed
-
- Akiyama M, Sawamura D, Nomura Y, Sugawara M, Shimizu H. Truncation of CGI-58 protein causes malformation of lamellar granules resulting in ichthyosis in Dorfman-Chanarin syndrome. J Invest Dermatol. 2003;121(5):1029–34. - PubMed
-
- Bruno C, Bertini E, Di Rocco M, Cassandrini D, Ruffa G, De Toni T, et al. Clinical and genetic characterization of Chanarin-Dorfman syndrome. Biochem Biophys Res Commun. 2008;369(4):1125–8. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
