

LMFE: A Novel Method for Predicting Plant LncRNA Based on Multi-Feature Fusion and Ensemble Learning
- PMID: 40282384
- PMCID: PMC12026654
- DOI: 10.3390/genes16040424
LMFE: A Novel Method for Predicting Plant LncRNA Based on Multi-Feature Fusion and Ensemble Learning
Abstract
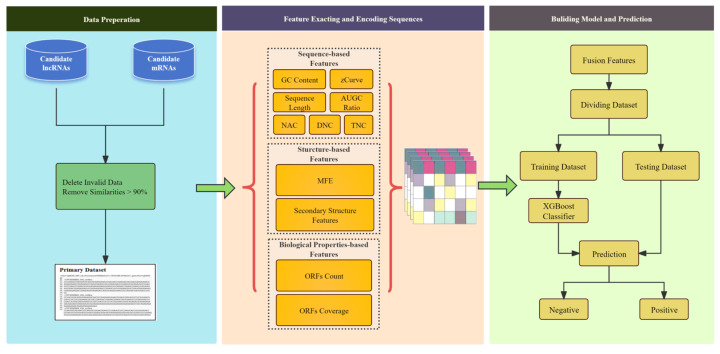
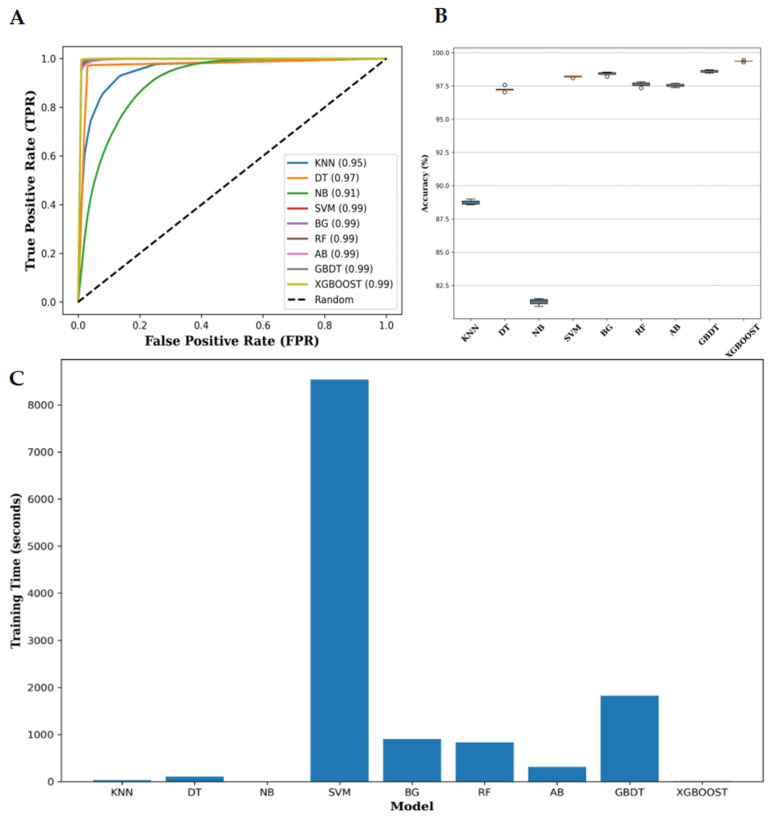
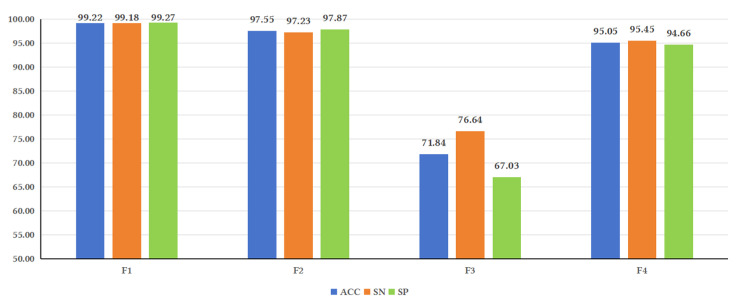
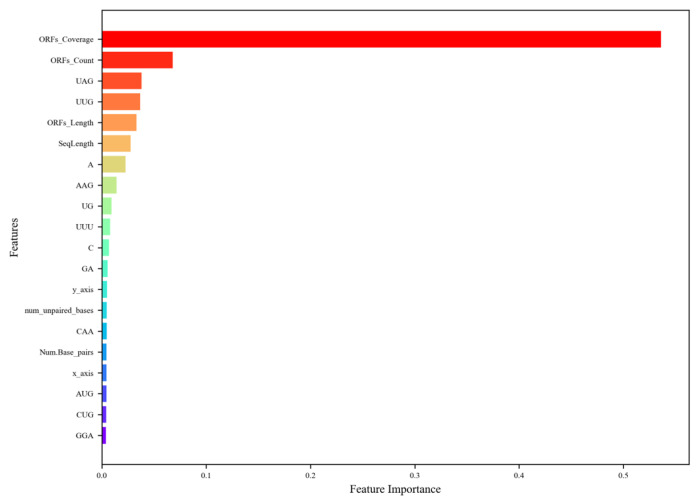
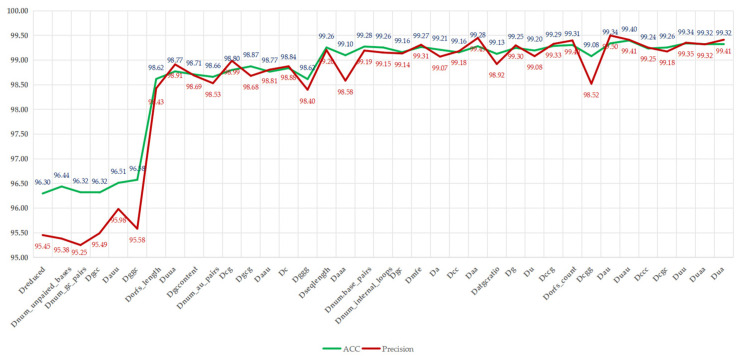
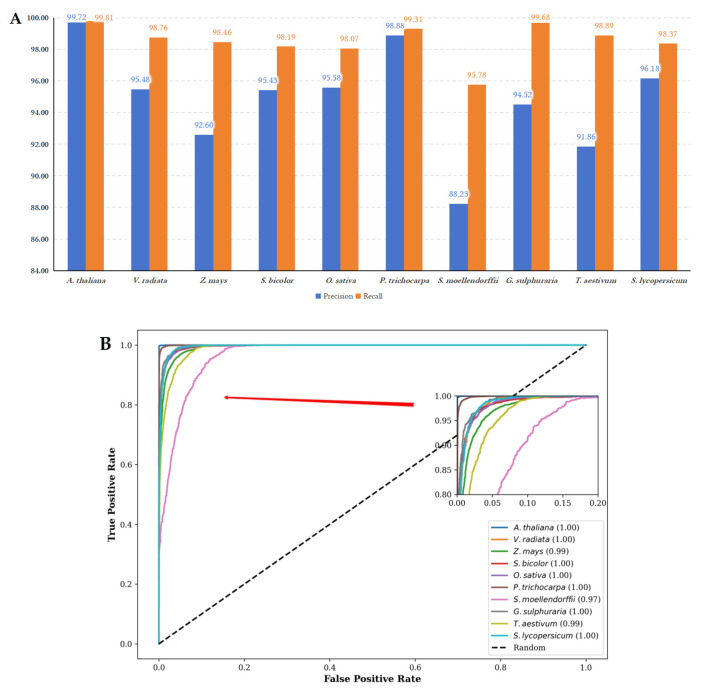
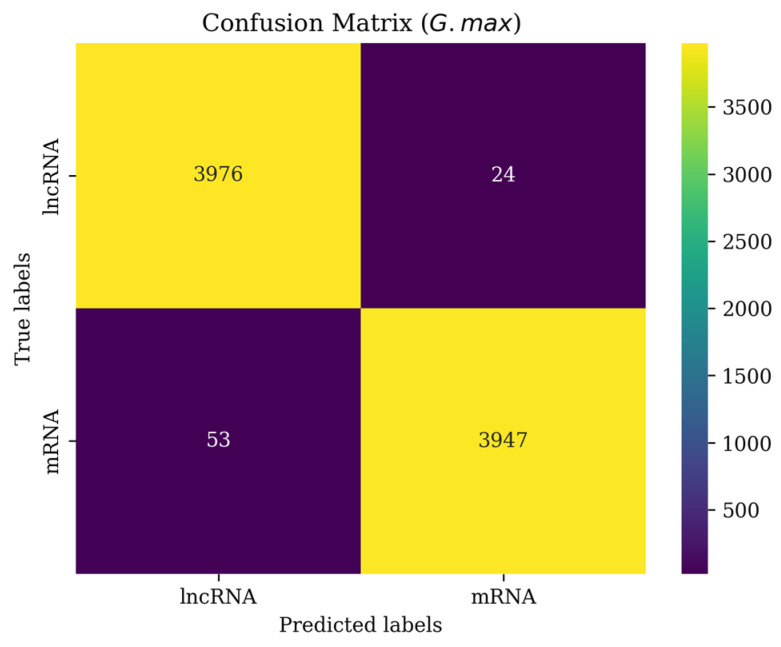
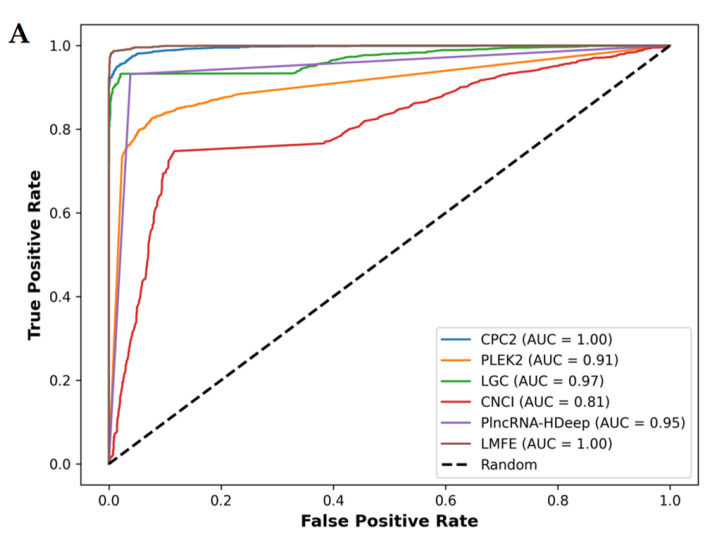
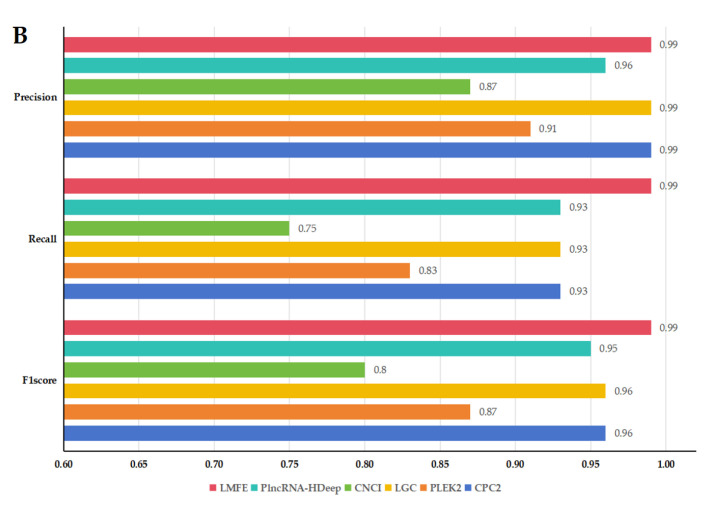
Background/Objectives: Long non-coding RNAs (lncRNAs) play a crucial regulatory role in plant trait expression and disease management, making their accurate prediction a key research focus for guiding biological experiments. While extensive studies have been conducted on animals and humans, plant lncRNA research remains relatively limited due to various challenges, such as data scarcity and genomic complexity. This study aims to bridge this gap by developing an effective computational method for predicting plant lncRNAs, specifically by classifying transcribed RNA sequences as lncRNAs or mRNAs using multi-feature analysis. Methods: We propose the lncRNA multi-feature-fusion ensemble learning (LMFE) approach, a novel method that integrates 100-dimensional features from RNA biological properties-based, sequence-based, and structure-based features, employing the XGBoost ensemble learning algorithm for prediction. To address unbalanced datasets, we implemented the synthetic minority oversampling technique (SMOTE). LMFE was validated across benchmark datasets, cross-species datasets, unbalanced datasets, and independent datasets. Results: LMFE achieved an accuracy of 99.42%, an F1score of 0.99, and an MCC of 0.98 on the benchmark dataset, with robust cross-species performance (accuracy ranging from 89.30% to 99.81%). On unbalanced datasets, LMFE attained an average accuracy of 99.41%, representing a 12.29% improvement over traditional methods without SMOTE (average ACC of 87.12%). Compared to state-of-the-art methods, such as CPC2 and PLEKv2, LMFE consistently outperformed them across multiple metrics on independent datasets (with an accuracy ranging from 97.33% to 99.21%), with redundant features having minimal impact on performance. Conclusions: LMFE provides a highly accurate and generalizable solution for plant lncRNA prediction, outperforming existing methods through multi-feature fusion and ensemble learning while demonstrating robustness to redundant features. Despite its effectiveness, variations in performance across species highlight the necessity for future improvements in managing diverse plant genomes. This method represents a valuable tool for advancing plant lncRNA research and guiding biological experiments.
Keywords: LMFE; cross-species; ensemble learning; multi-feature fusion; plant lncRNA prediction.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Gauthier J., Vincent A.T., Charette S.J., Derome N. A brief history of bioinformatics. Brief. Bioinform. 2019;20:1981–1996. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
