

Deciphering Gut Microbiome in Colorectal Cancer via Robust Learning Methods
- PMID: 40282413
- PMCID: PMC12026925
- DOI: 10.3390/genes16040452
Deciphering Gut Microbiome in Colorectal Cancer via Robust Learning Methods
Abstract
Background: Colorectal cancer (CRC) is one of the most prevalent cancers worldwide and is closely linked to the gut microbiota. Identifying reproducible and generalizable microbial signatures holds significant potential for enhancing early detection and advancing treatment for this deadly disease.
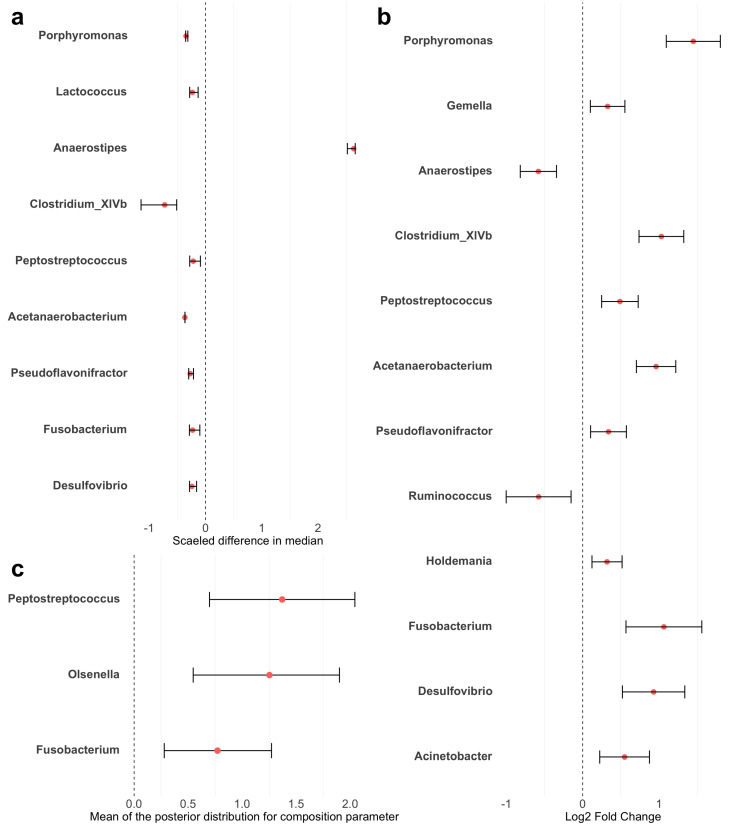
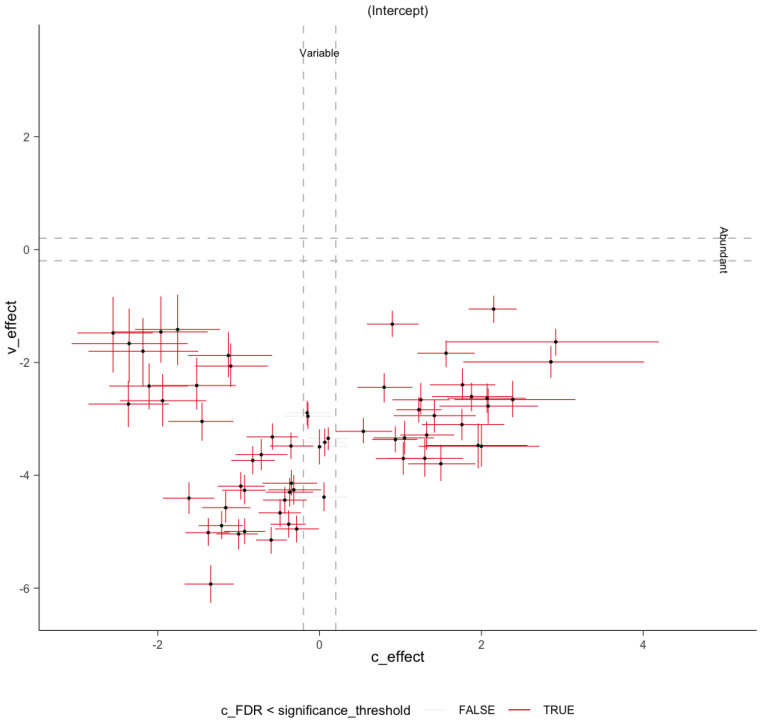
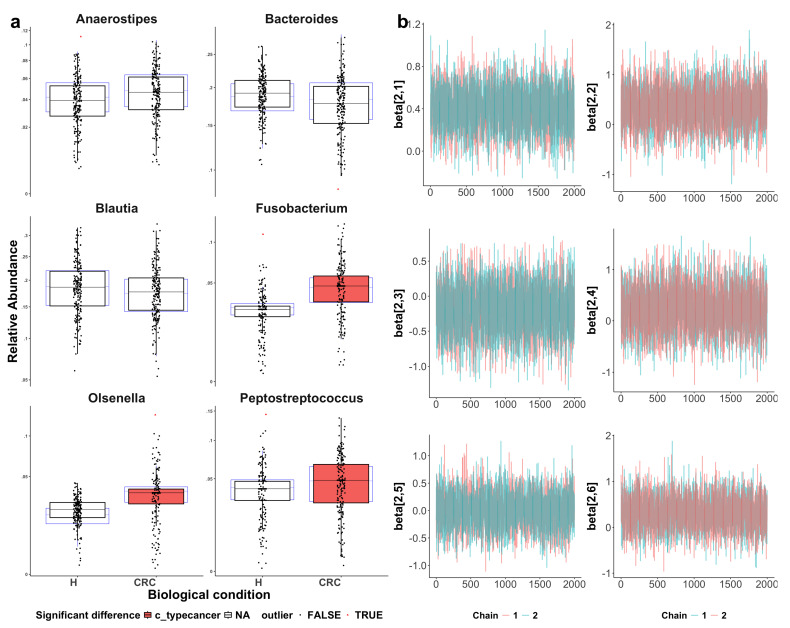
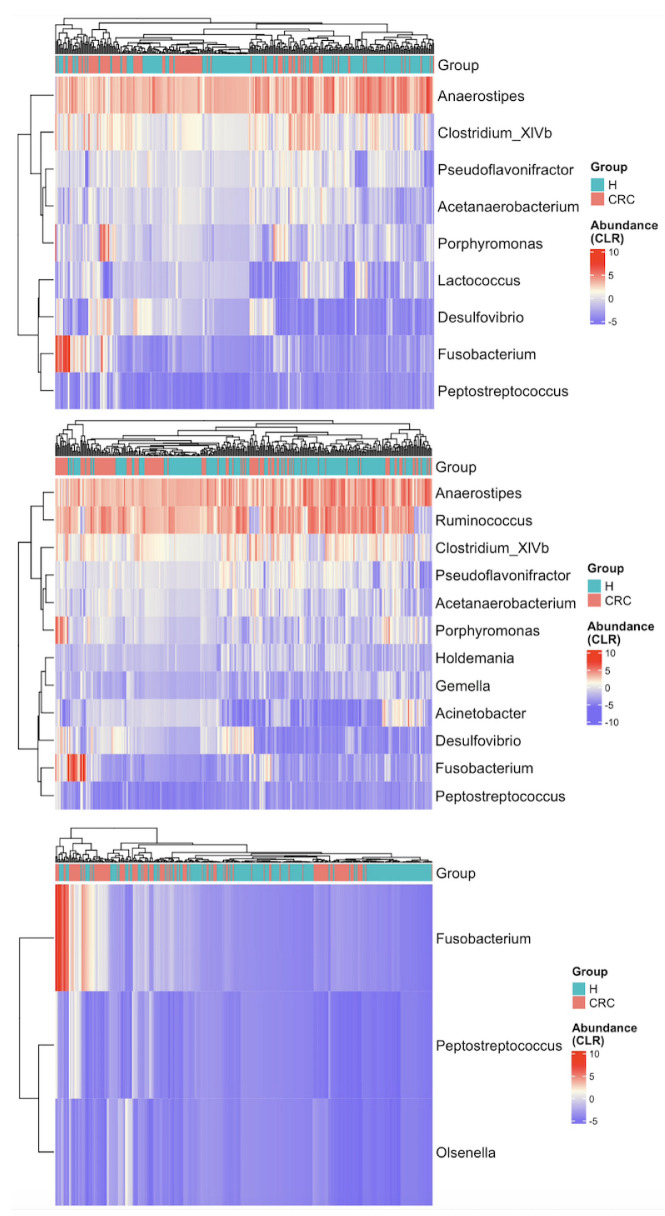
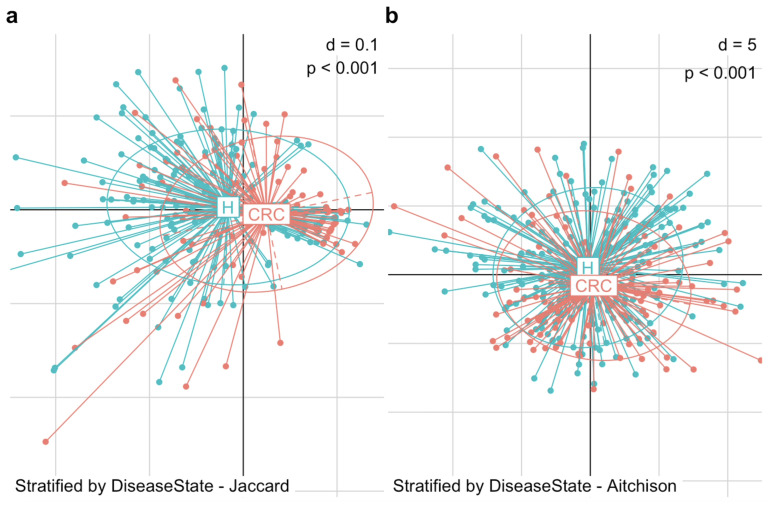
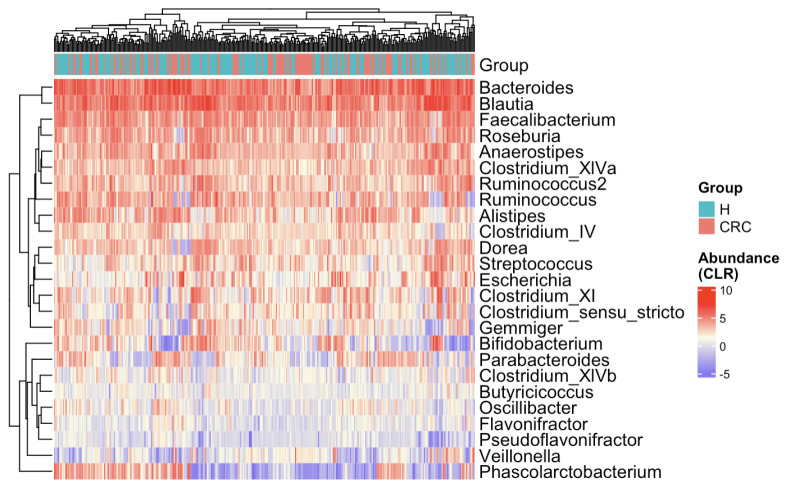
Methods: This study integrated various publicly available case-control datasets to identify microbial signatures for CRC. Alpha and beta diversity metrics were evaluated to characterize differences in gut microbial richness, evenness, and overall composition between CRC patients and healthy controls. Differential abundance analysis was conducted using ANCOM-BC and LEfSe to pinpoint individual taxa that were enriched or depleted in CRC patients. Additionally, sccomp, a Bayesian machine learning method from single-cell analysis, was adapted to provide a more robust validation of compositional differences in individual microbial markers.
Results: Gut microbial richness is significantly higher in CRC patients, and overall microbiome composition differs significantly between CRC patients and healthy controls. Several taxa, such as Fusobacterium and Peptostreptococcus, are enriched in CRC patients, while others, including Anaerostipes, are depleted. The microbial signatures identified from the integrated data are reproducible and generalizable, with many aligning with findings from previous studies. Furthermore, the use of sccomp enhanced the precision of individual microbial marker identification.
Conclusions: Biologically, the microbial signatures identified from the integrated data improve our understanding of the gut microbiota's role in CRC pathogenesis and may contribute to the development of translational targets and microbiota-based therapies. Methodologically, this study demonstrates the effectiveness of adapting robust techniques from single-cell research to improve the precision of microbial marker discovery.
Keywords: colorectal cancer; data integration; gut microbiota; microbial signatures; robust differential composition analysis.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures



Similar articles
-
Meta-analysis of gut microbiome reveals patterns of dysbiosis in colorectal cancer patients.J Med Microbiol. 2025 Jul;74(7):002042. doi: 10.1099/jmm.0.002042. J Med Microbiol. 2025. PMID: 40737178 Free PMC article.
-
The characteristics of tissue microbiota in different anatomical locations and different tissue types of the colorectum in patients with colorectal cancer.mSystems. 2025 Jun 17;10(6):e0019825. doi: 10.1128/msystems.00198-25. Epub 2025 May 27. mSystems. 2025. PMID: 40422085 Free PMC article.
-
Exploring the gut microbiota associated with peripheral nerve invasion in colorectal cancer patients and constructing predictive models.BMC Microbiol. 2025 Aug 16;25(1):514. doi: 10.1186/s12866-025-04179-x. BMC Microbiol. 2025. PMID: 40818935 Free PMC article.
-
Efficacy and safety of herbal formulas with the function of gut microbiota regulation for gastric and colorectal cancer: A systematic review and meta-analysis.Front Cell Infect Microbiol. 2022 Aug 4;12:875225. doi: 10.3389/fcimb.2022.875225. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 35992176 Free PMC article.
-
Contrast-enhanced ultrasound using SonoVue® (sulphur hexafluoride microbubbles) compared with contrast-enhanced computed tomography and contrast-enhanced magnetic resonance imaging for the characterisation of focal liver lesions and detection of liver metastases: a systematic review and cost-effectiveness analysis.Health Technol Assess. 2013 Apr;17(16):1-243. doi: 10.3310/hta17160. Health Technol Assess. 2013. PMID: 23611316 Free PMC article.
References
-
- Yachida S., Mizutani S., Shiroma H., Shiba S., Nakajima T., Sakamoto T., Watanabe H., Masuda K., Nishimoto Y., Kubo M., et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019;25:968–976. doi: 10.1038/s41591-019-0458-7. - DOI - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
