

Urea cycle defects in adulthood: clinical presentation, diagnosis and treatment in genetically encoded hepatic metabolic disorders with a potential for encephalopathy
- PMID: 40285952
- PMCID: PMC12033206
- DOI: 10.1007/s11011-025-01619-5
Urea cycle defects in adulthood: clinical presentation, diagnosis and treatment in genetically encoded hepatic metabolic disorders with a potential for encephalopathy
Abstract
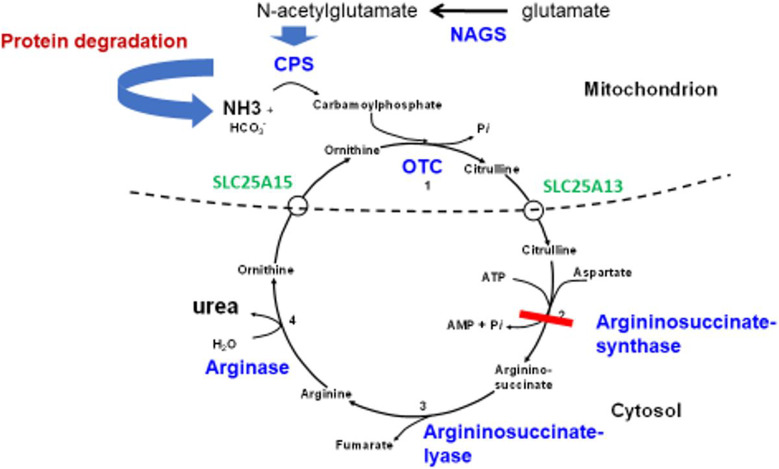
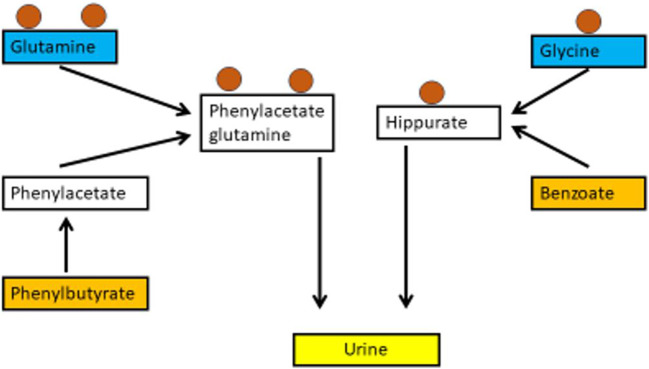
Hyperammonaemia is an important cause for encephalopathy. Ammonia is the waste product of amino acid degradation and cannot be excreted via urine. Ammonia is metabolized to water-soluble urea via the urea cycle. Hyperammonaemia not only occurs during acute liver failure, but also in rare genetically determined defects of enzymes or transporters involved in the urea cycle resulting in elevated ammonia concentrations. Enzyme defects include deficiency of carbamylphosphate synthase, N-acetylglutamate synthase, ornithine transcarbamylase, argininosuccinate lyase and arginase, transporter defects are citrin deficiency and HHH-syndrome. These urea cycle defects (UCD) mostly manifest for the first time during the neonatal period, infancy or childhood, however first clinical manifestations including encephalopathy may be observed in adulthood in milder forms. Therefore, physicians treating adults should be aware of clinical symptoms in UCD to make a timely diagnosis and initiate treatment. In adulthood, clinical symptoms are often uncharacteristic including headache, avoidance of high-protein food, psychiatric symptoms triggered by heavy exercise or delivery of a child, autism, attention deficit, lethargy, developmental delay and epilepsy. Elevated ammonia concentrations in blood are the biochemical hallmark. Some UCDs can be diagnosed at metabolite level, others only at genetic level. Treatment consists of eucaloric, low-protein diet supplemented with essential amino acids and vitamins/trace elements, and intake of arginine or citrulline. Pharmacological scavengers of nitrogen are benzoate and butyrate. If conservative therapy fails, hemodialysis should be considered. Prompt treatment during acute crises is essential for optimal outcome. Liver transplantation is considered in metabolically unstable patients. For arginase deficiency, enzyme replacement therapy is available.
Keywords: Amino acids; Ammonia; Benzoate; Glycerophenylbutyrate; Scavengers; Urea cycle defect.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
The hyperornithinemia-hyperammonemia-homocitrullinuria syndrome.Orphanet J Rare Dis. 2015 Mar 11;10:29. doi: 10.1186/s13023-015-0242-9. Orphanet J Rare Dis. 2015. PMID: 25874378 Free PMC article.
-
Developing splice-switching oligonucleotides for urea cycle disorder using an integrated diagnostic and therapeutic platform.J Hepatol. 2025 Aug;83(2):411-425. doi: 10.1016/j.jhep.2025.02.007. Epub 2025 Feb 18. J Hepatol. 2025. PMID: 39978599
-
AGA Clinical Practice Update on GI Manifestations and Autonomic or Immune Dysfunction in Hypermobile Ehlers-Danlos Syndrome: Expert Review.Clin Gastroenterol Hepatol. 2025 Jul;23(8):1291-1302. doi: 10.1016/j.cgh.2025.02.015. Epub 2025 May 19. Clin Gastroenterol Hepatol. 2025. PMID: 40387691 Review.
-
Apert Syndrome.2019 May 30. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2019 May 30. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 31145570 Free Books & Documents. Review.
-
The current social status in adult patients with urea cycle disorders in Japan.Mol Genet Metab. 2025 Aug;145(4):109185. doi: 10.1016/j.ymgme.2025.109185. Epub 2025 Jul 1. Mol Genet Metab. 2025. PMID: 40618446
References
-
- Ah Mew N, Simpson KL, Gropman AL, Lanpher BC, Chapman KA, Summar ML (2003 Apr 29 [updated 2017 Jun 22]) Urea cycle disorders overview. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A (eds). GeneReviews® [Internet]. University of Washington, Seattle, Seattle, pp 1993–2024
-
- Ah Mew N, McCarter R, Izem R, Markus A, Gerstein M, Rice K, Sanz J, Le Mons C, Bartos J, Tuchman M (2020) Comparing treatment options for urea cycle disorders [Internet]. Washington (DC): Patient-Centered Outcomes Research Institute (PCORI) - PubMed
-
- Aires CC, van Cruchten A, Ijlst L, de Almeida IT, Duran M, Wanders RJ, Silva MF (2011) New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol 55(2):426–434. 10.1016/j.jhep.2010.11.031 - PubMed
-
- Amayreh W, Meyer U, Das AM (2014) Treatment of arginase deficiency revisited: guanidinoacetate as a therapeutic target and biomarker for therapeutic monitoring. Dev Med Child Neurol 56(10):1021–1024. 10.1111/dmcn.12488 - PubMed
-
- Baruteau J, Jameson E, Morris AA, Chakrapani A, Santra S, Vijay S, Kocadag H, Beesley CE, Grunewald S, Murphy E, Cleary M, Mundy H, Abulhoul L, Broomfield A, Lachmann R, Rahman Y, Robinson PH, MacPherson L, Foster K, Chong WK, Ridout DA, Bounford KM, Waddington SN, Mills PB, Gissen P, Davison JE (2017) Expanding the phenotype in argininosuccinic aciduria: need for new therapies. J Inherit Metab Dis 40(3):357–368. 10.1007/s10545-017-0022-x - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
