

Molecular mechanisms and targeted therapy of progranulin in metabolic diseases
- PMID: 40290306
- PMCID: PMC12021630
- DOI: 10.3389/fendo.2025.1553794
Molecular mechanisms and targeted therapy of progranulin in metabolic diseases
Abstract
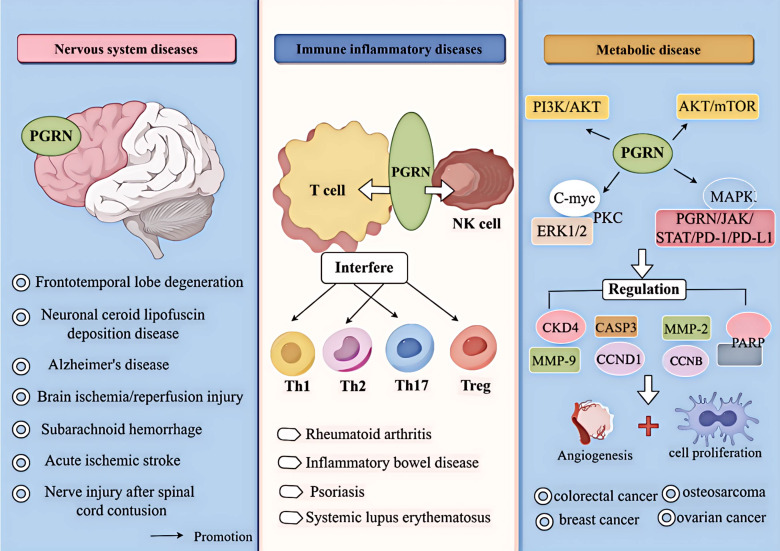
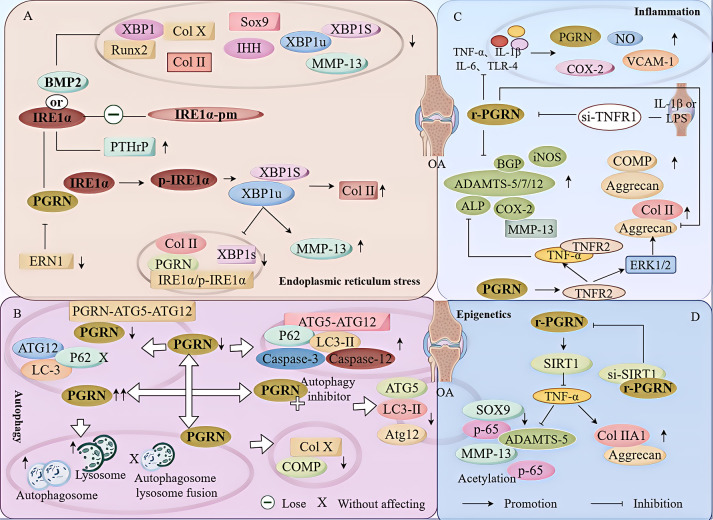
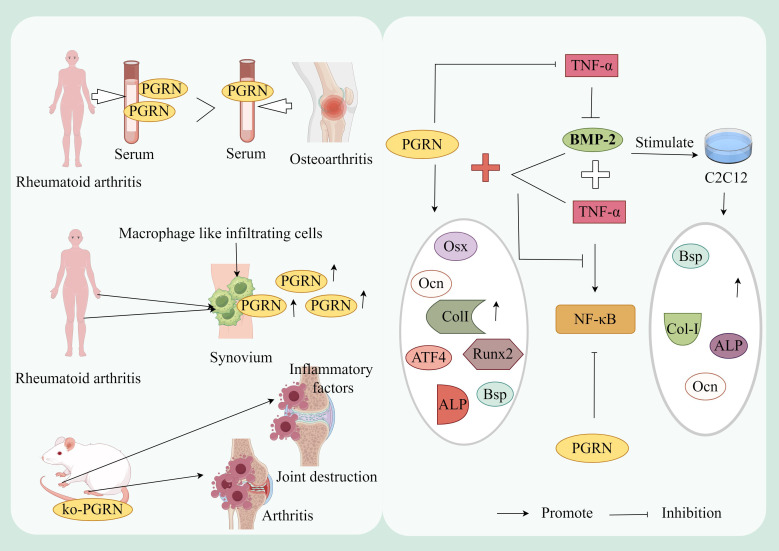
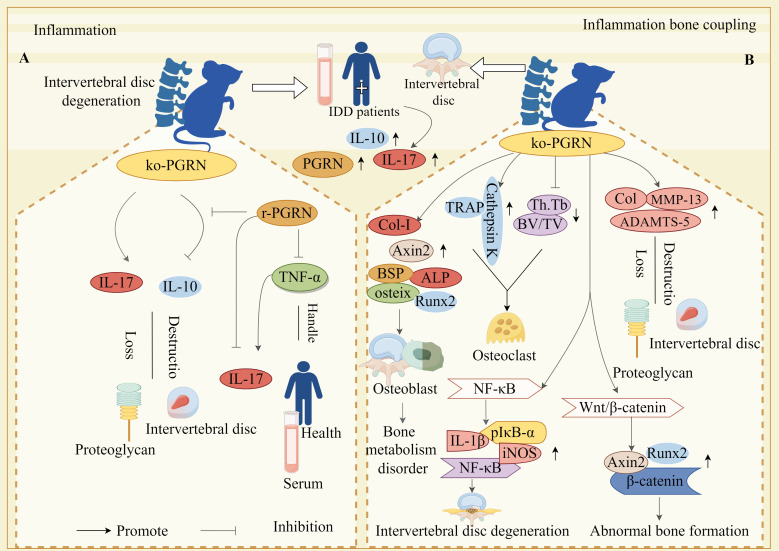
Progranulin (PGRN) is a secreted glycoprotein with cytokine-like properties, exerting tripartite mechanisms of inflammation suppression, tissue repair promotion, and metabolic regulation. This multifaceted functionality positions PGRN as a potential "multi-effect therapeutic strategy" for metabolic disorders characterised by cartilage degradation and imbalanced bone remodelling, potentially establishing it as a novel therapeutic target for such conditions. Osteoarthritis, rheumatoid arthritis, intervertebral disc degeneration, osteoporosis, periodontitis, and diabetes-related complications-representing the most prevalent metabolic diseases-currently lack effective treatments due to incomplete understanding of their precise pathogenic mechanisms. Recent studies have revealed that PGRN expression levels are closely associated with the onset and progression of these metabolic disorders. However, the exact regulatory role of PGRN in these diseases remains elusive, partly owing to its tissue-specific actions and context-dependent dual roles (anti-inflammatory vs. pro-inflammatory). In this review, we summarise the structure and functions of PGRN, explore its involvement in neurological disorders, immune-inflammatory diseases, and metabolic conditions, and specifically focus on its molecular mechanisms in metabolic diseases. Furthermore, we consolidate advances in targeting PGRN and the application of its engineered derivative, Atsttrin, in metabolic bone disorders. We also discuss potential unexplored mechanisms through which PGRN may exert influence within this field or other therapeutic domains. Collectively, this work aims to provide a new framework for elucidating PGRN's role in disease pathogenesis and advancing strategies for the prevention and treatment of metabolic disorders.
Keywords: PGRN; bone homeostasis; cartilage repair; inflammation; metabolic diseases; targeted therapy.
Copyright © 2025 Wang, Liang, Yang, Shi, Shao, Jing, Yang, Chu, An, Zhou, Song, Chen and Liu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Progranulin is essential for bone homeostasis and immunology.Ann N Y Acad Sci. 2022 Dec;1518(1):58-68. doi: 10.1111/nyas.14905. Epub 2022 Sep 30. Ann N Y Acad Sci. 2022. PMID: 36177883 Review.
-
Progranulin in Musculoskeletal Inflammatory and Degenerative Disorders, Focus on Rheumatoid Arthritis, Lupus and Intervertebral Disc Disease: A Systematic Review.Pharmaceuticals (Basel). 2022 Dec 12;15(12):1544. doi: 10.3390/ph15121544. Pharmaceuticals (Basel). 2022. PMID: 36558994 Free PMC article. Review.
-
Progranulin deficiency exacerbates spinal cord injury by promoting neuroinflammation and cell apoptosis in mice.J Neuroinflammation. 2019 Nov 27;16(1):238. doi: 10.1186/s12974-019-1630-1. J Neuroinflammation. 2019. PMID: 31775776 Free PMC article.
-
Progranulinopathy: A diverse realm of disorders linked to progranulin imbalances.Cytokine Growth Factor Rev. 2024 Apr;76:142-159. doi: 10.1016/j.cytogfr.2023.11.001. Epub 2023 Nov 11. Cytokine Growth Factor Rev. 2024. PMID: 37981505 Free PMC article. Review.
-
Microglial Progranulin: Involvement in Alzheimer's Disease and Neurodegenerative Diseases.Cells. 2019 Mar 11;8(3):230. doi: 10.3390/cells8030230. Cells. 2019. PMID: 30862089 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous