

Aminoacyl-tRNA synthetase urzymes optimized by deep learning behave as a quasispecies
- PMID: 40290414
- PMCID: PMC12033045
- DOI: 10.1063/4.0000294
Aminoacyl-tRNA synthetase urzymes optimized by deep learning behave as a quasispecies
Abstract
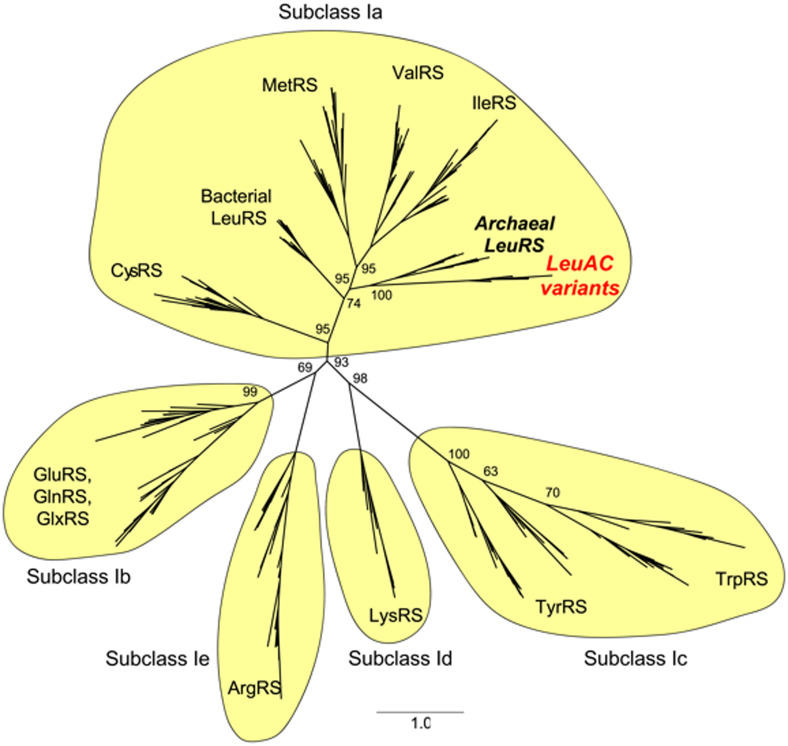
Protein design plays a key role in our efforts to work out how genetic coding began. That effort entails urzymes. Urzymes are small, conserved excerpts from full-length aminoacyl-tRNA synthetases that remain active. Urzymes require design to connect disjoint pieces and repair naked nonpolar patches created by removing large domains. Rosetta allowed us to create the first urzymes, but those urzymes were only sparingly soluble. We could measure activity, but it was hard to concentrate those samples to levels required for structural biology. Here, we used the deep learning algorithms ProteinMPNN and AlphaFold2 to redesign a set of optimized LeuAC urzymes derived from leucyl-tRNA synthetase. We select a balanced, representative subset of eight variants for testing using principal component analysis. Most tested variants are much more soluble than the original LeuAC. They also span a range of catalytic proficiency and amino acid specificity. The data enable detailed statistical analyses of the sources of both solubility and specificity. In that way, we show how to begin to unwrap the elements of protein chemistry that were hidden within the neural networks. Deep learning networks have thus helped us surmount several vexing obstacles to further investigations into the nature of ancestral proteins. Finally, we discuss how the eight variants might resemble a sample drawn from a population similar to one subject to natural selection.
© 2025 Author(s).
Conflict of interest statement
The authors have no conflicts to disclose.
Figures









Similar articles
-
A Leucyl-tRNA Synthetase Urzyme: Authenticity of tRNA Synthetase Catalytic Activities and Promiscuous Phosphorylation of Leucyl-5'AMP.Int J Mol Sci. 2022 Apr 11;23(8):4229. doi: 10.3390/ijms23084229. Int J Mol Sci. 2022. PMID: 35457045 Free PMC article.
-
A genomic database furnishes minimal functional glycyl-tRNA synthetases homologous to other, designed class II urzymes.Nucleic Acids Res. 2024 Nov 27;52(21):13305-13324. doi: 10.1093/nar/gkae992. Nucleic Acids Res. 2024. PMID: 39494520 Free PMC article.
-
Domain acquisition by class I aminoacyl-tRNA synthetase urzymes coordinated the catalytic functions of HVGH and KMSKS motifs.Nucleic Acids Res. 2023 Aug 25;51(15):8070-8084. doi: 10.1093/nar/gkad590. Nucleic Acids Res. 2023. PMID: 37470821 Free PMC article.
-
Class I and II aminoacyl-tRNA synthetase tRNA groove discrimination created the first synthetase-tRNA cognate pairs and was therefore essential to the origin of genetic coding.IUBMB Life. 2019 Aug;71(8):1088-1098. doi: 10.1002/iub.2094. Epub 2019 Jun 13. IUBMB Life. 2019. PMID: 31190358 Free PMC article. Review.
-
Coding of Class I and II Aminoacyl-tRNA Synthetases.Adv Exp Med Biol. 2017;966:103-148. doi: 10.1007/5584_2017_93. Adv Exp Med Biol. 2017. PMID: 28828732 Free PMC article. Review.
Cited by
-
Structural Enzymology, Phylogenetics, Differentiation, and Symbolic Reflexivity at the Dawn of Biology.Genome Biol Evol. 2025 May 30;17(6):evaf095. doi: 10.1093/gbe/evaf095. Genome Biol Evol. 2025. PMID: 40476352 Free PMC article.
References
-
- Abramson J., Adler J., Dunger J., Evans R., Green T., Pritzel A., Ronneberger O., Willmore L., Ballard A. J., Bambrick J., Bodenstein S. W., Evans D. A., Hung C.-C., O'Neill M., Reiman D., Tunyasuvunakool K., Wu Z., Žemgulytė A., Arvaniti E., Beattie C., Bertolli O., Bridgland A., Cherepanov A., Congreve M., Cowen-Rivers A. I., Cowie A., Figurnov M., Fuchs F. B., Gladman H., Jain R., Khan Y. A., Low C. M. R., Perlin K., Potapenko A., Savy P., Singh S., Stecula A., Thillaisundaram A., Tong C., Yakneen S., Zhong E. D., Zielinski M., Žídek A., Bapst V., Kohli P., Jaderberg M., Hassabis D., and Jumper J. M., “Accurate structure prediction of biomolecular interactions with AlphaFold 3,” Nature 630, 493 (2024).10.1038/s41586-024-07487-w - DOI - PMC - PubMed
-
- Dauparas J., Anishchenko I., Bennett N., Bai H., Ragotte R. J., Milles L. F., Wicky B. I. M., Courbet A., de Haas R. J., Bethel N., Leung P. J. Y., Huddy T. F., Pellock S., Tischer D., Chan F., Koepnick B., Nguyen H., Kang A., Sankaran B., Bera A. K., King N. P., and Baker D., “Robust deep learning-based protein sequence design using ProteinMPNN,” Science 378, 49–56 (2022).10.1126/science.add2187 - DOI - PMC - PubMed
-
- Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., Bridgland A., Meyer C., Kohl S. A. A., Ballard A. J., Cowie A. T., Romera-Paredes B., Nikolov S., Jain R., Adler J., Back T., Petersen S., Reiman D., Clancy E., Zielinski M., Steinegger M., Pacholska M., Berghammer T., Bodenstein S., Silver D., Vinyals O., Senior A. W., Kavukcuoglu K., Kohli P., and Hassabis D., “Highly accurate protein structure prediction with AlphaFold,” Nature 596, 583–592 (2021).10.1038/s41586-021-03819-2 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources