

Mechanisms underlying targeted mitochondrial therapy for programmed cardiac cell death
- PMID: 40292006
- PMCID: PMC12021874
- DOI: 10.3389/fphys.2025.1548194
Mechanisms underlying targeted mitochondrial therapy for programmed cardiac cell death
Abstract
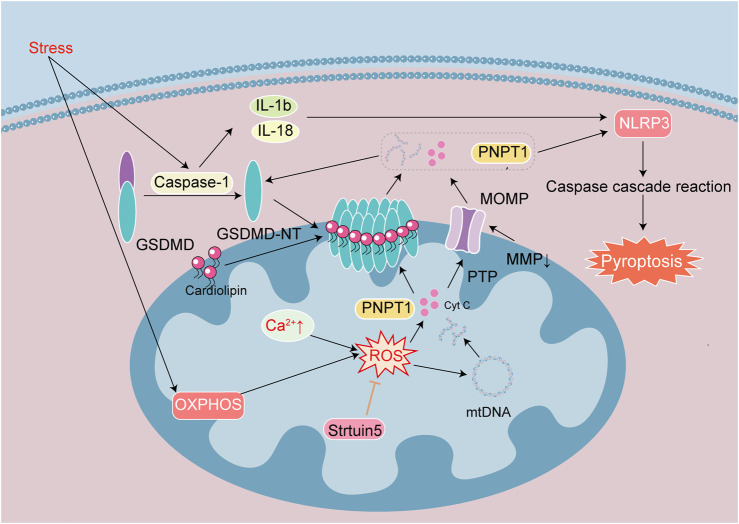
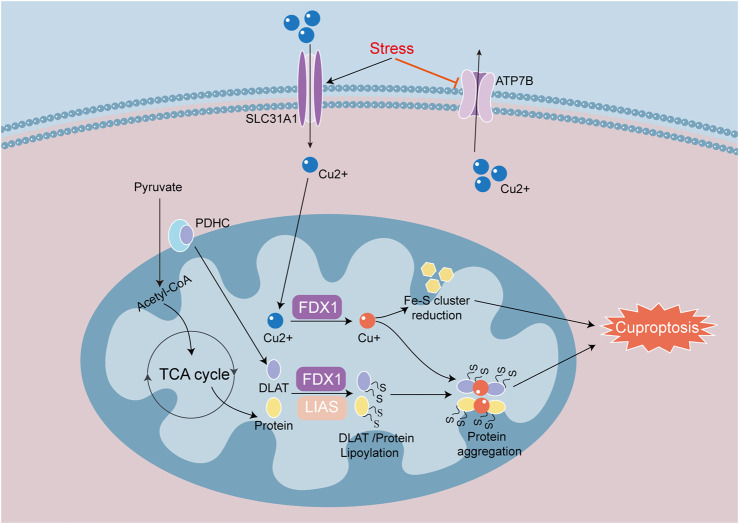
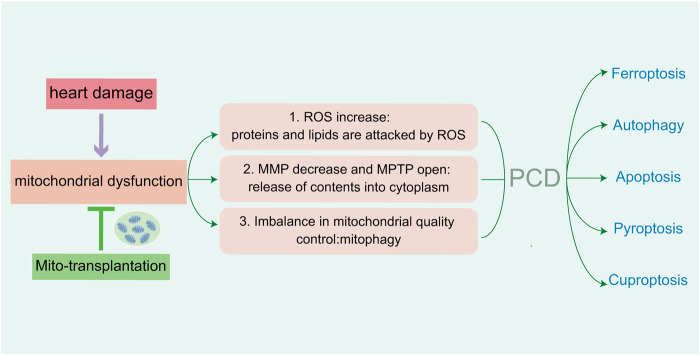
Heart diseases are common clinical diseases, such as cardiac fibrosis, heart failure, hypertension and arrhythmia. Globally, the incidence rate and mortality of heart diseases are increasing by years. The main mechanism of heart disease is related to the cellular state. Mitochondrion is the organ of cellular energy supply, participating in various signal transduction pathways and playing a vital role in the occurrence and development of heart disease. This review summarizes the cell death patterns and molecular mechanisms associated with heart disease and mitochondrial dysfunction.
Keywords: PCD; apoptosis; autophagy; cardiomyocyte; cuproptosis; ferroptosis; mitochondria; pyroptosis.
Copyright © 2025 Jing, Zhao, Xiong, Zeng, Jiang and Li.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease.Physiol Rev. 2019 Oct 1;99(4):1765-1817. doi: 10.1152/physrev.00022.2018. Physiol Rev. 2019. PMID: 31364924 Free PMC article. Review.
-
Autophagy, Ferroptosis, Apoptosis and Pyroptosis in Metabolic Dysfunction-Associated Steatotic Liver Disease.Front Biosci (Landmark Ed). 2024 Jan 19;29(1):30. doi: 10.31083/j.fbl2901030. Front Biosci (Landmark Ed). 2024. PMID: 38287834 Review.
-
m6A control programmed cell death in cardiac fibrosis.Life Sci. 2024 Sep 15;353:122922. doi: 10.1016/j.lfs.2024.122922. Epub 2024 Jul 18. Life Sci. 2024. PMID: 39032691 Review.
-
Programmed Cell Death: Complex Regulatory Networks in Cardiovascular Disease.Front Cell Dev Biol. 2021 Nov 26;9:794879. doi: 10.3389/fcell.2021.794879. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34901035 Free PMC article. Review.
-
Emerging connectivity of programmed cell death pathways and pulmonary vascular remodelling during pulmonary hypertension.J Cell Mol Med. 2024 Aug;28(16):e70003. doi: 10.1111/jcmm.70003. J Cell Mol Med. 2024. PMID: 39153207 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources