

Variable rates of SARS-CoV-2 evolution in chronic infections
- PMID: 40294077
- PMCID: PMC12061394
- DOI: 10.1371/journal.ppat.1013109
Variable rates of SARS-CoV-2 evolution in chronic infections
Abstract
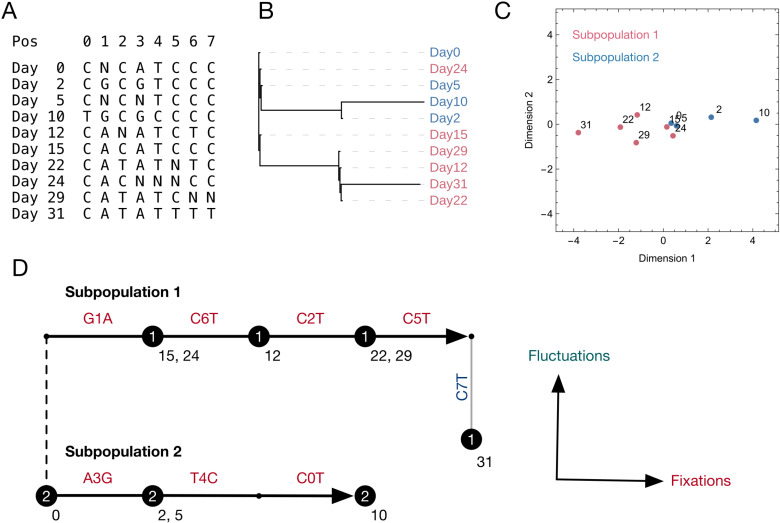
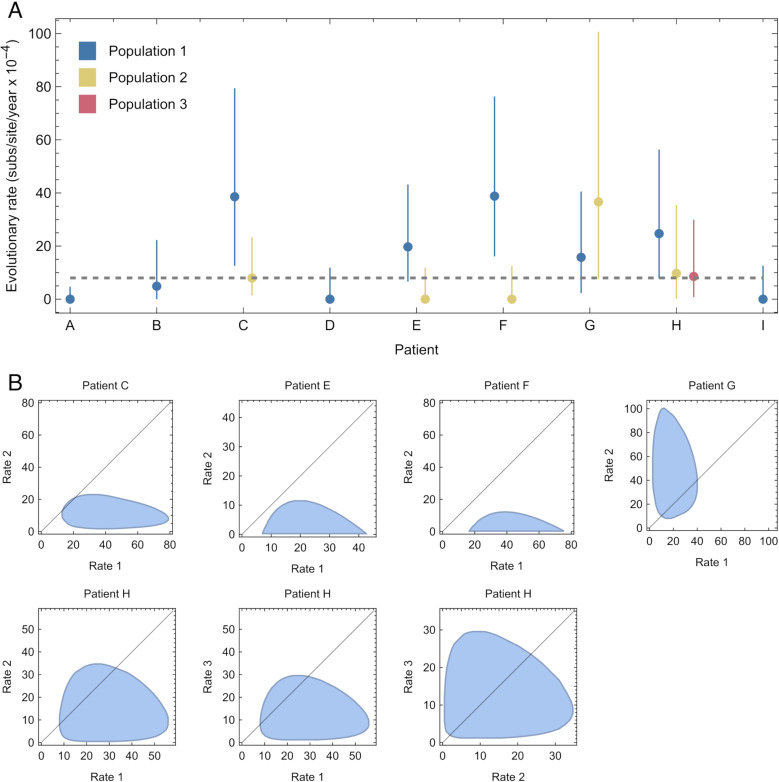
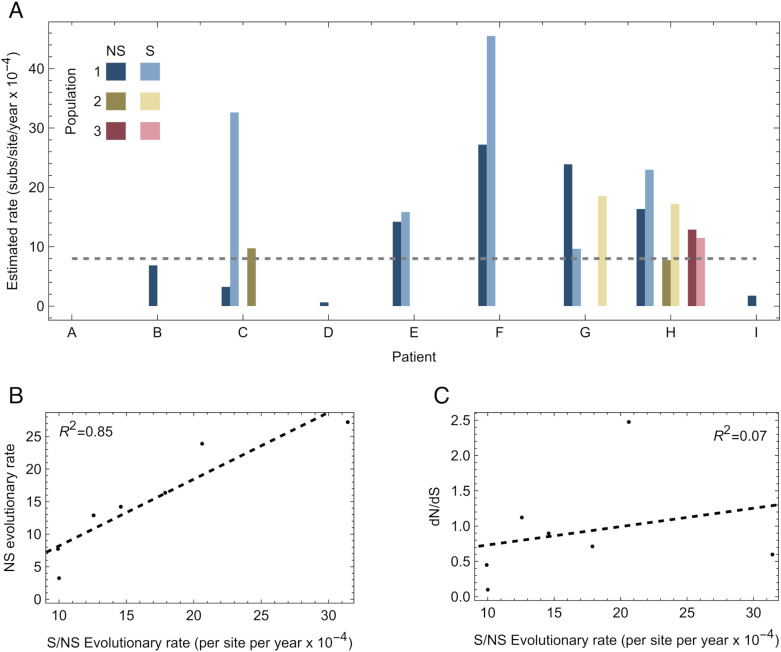
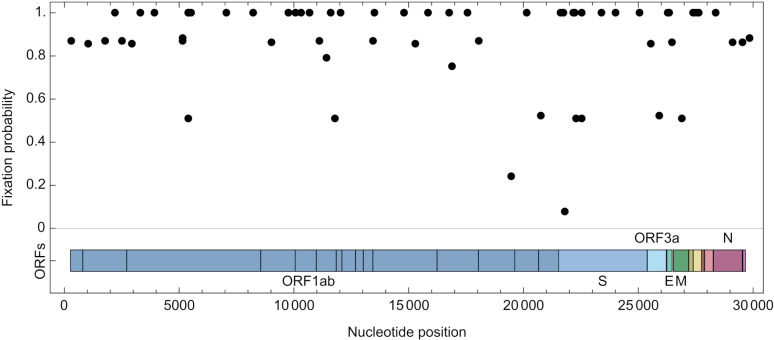
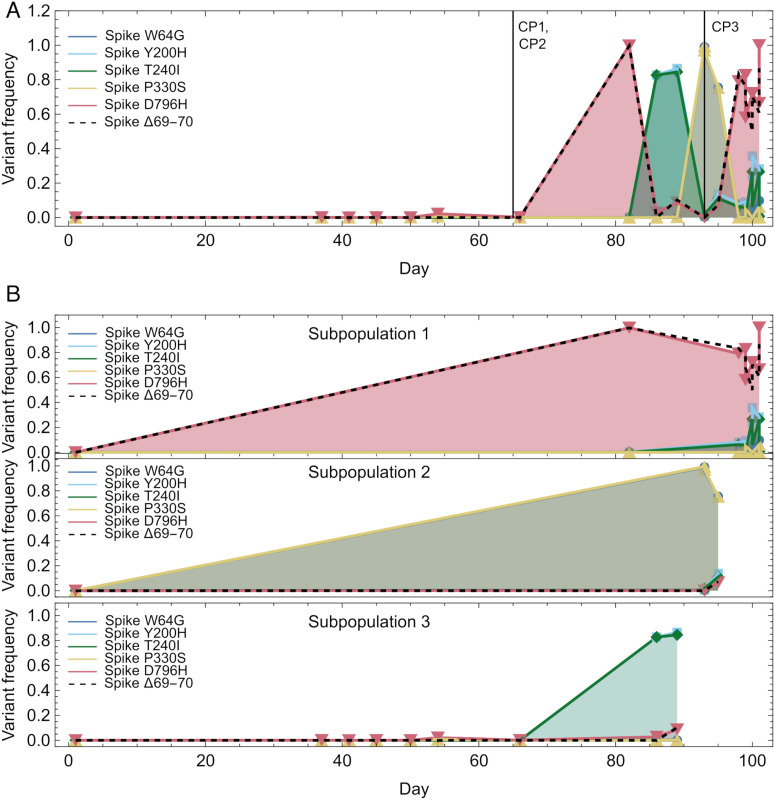
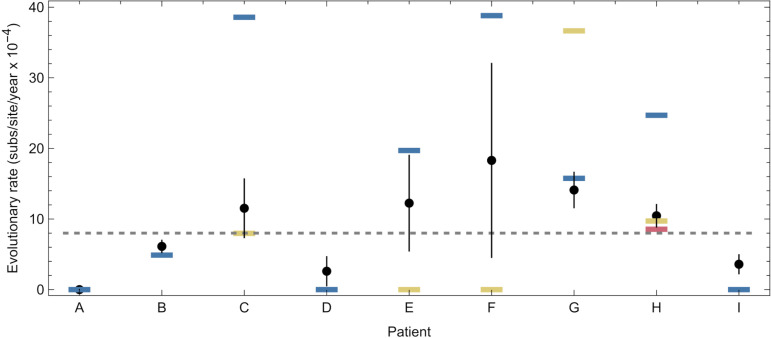
An important feature of the evolution of the SARS-CoV-2 virus has been the emergence of highly mutated novel variants, which are characterised by the gain of multiple mutations relative to viruses circulating in the general global population. Cases of chronic viral infection have been suggested as an explanation for this phenomenon, whereby an extended period of infection, with an increased rate of evolution, creates viruses with substantial genetic novelty. However, measuring a rate of evolution during chronic infection is made more difficult by the potential existence of compartmentalisation in the viral population, whereby the viruses in a host form distinct subpopulations. We here describe and apply a novel statistical method to study within-host virus evolution, identifying the minimum number of subpopulations required to explain sequence data observed from cases of chronic infection, and inferring rates for within-host viral evolution. Across nine cases of chronic SARS-CoV-2 infection in hospitalised patients we find that non-trivial population structure is relatively common, with five cases showing evidence of more than one viral population evolving independently within the host. The detection of non-trivial population structure was more common in severely immunocompromised individuals (p = 0.04, Fisher's Exact Test). We find cases of within-host evolution proceeding significantly faster, and significantly slower, than that of the global SARS-CoV-2 population, and of cases in which viral subpopulations in the same host have statistically distinguishable rates of evolution. Non-trivial population structure was associated with high rates of within-host evolution that were systematically underestimated by a more standard inference method.
Copyright: © 2025 Smith et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist
Figures

References
-
- Rambaut A, Loman NJ, Pybus O, Barclay WS, Barrett J, Carabelli AM, et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. 2020 Dec.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
