

Single-cell Transcriptional Atlas of Human Hematopoiesis Reveals Genetic and Hierarchy-Based Determinants of Aberrant AML Differentiation
- PMID: 40294241
- PMCID: PMC12209776
- DOI: 10.1158/2643-3230.BCD-24-0342
Single-cell Transcriptional Atlas of Human Hematopoiesis Reveals Genetic and Hierarchy-Based Determinants of Aberrant AML Differentiation
Abstract
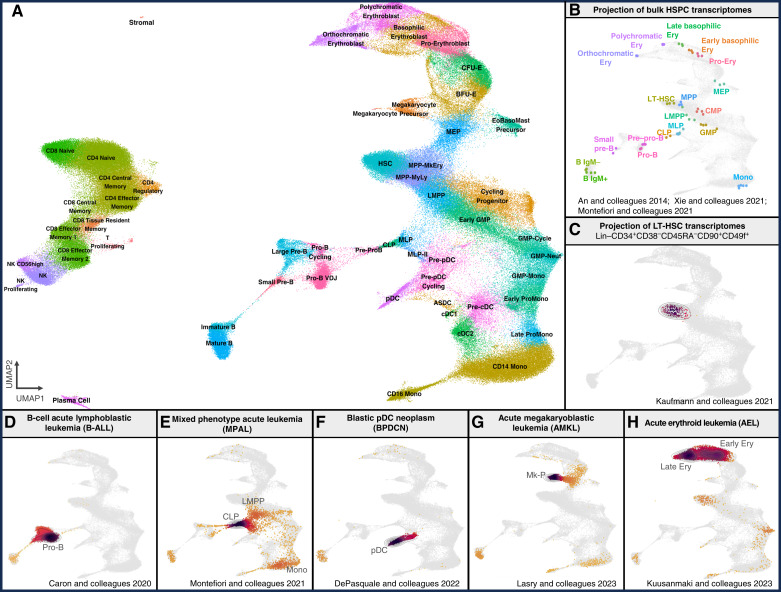
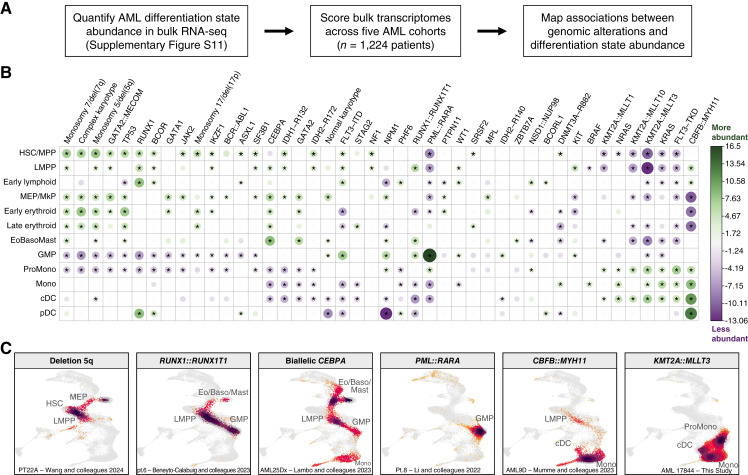
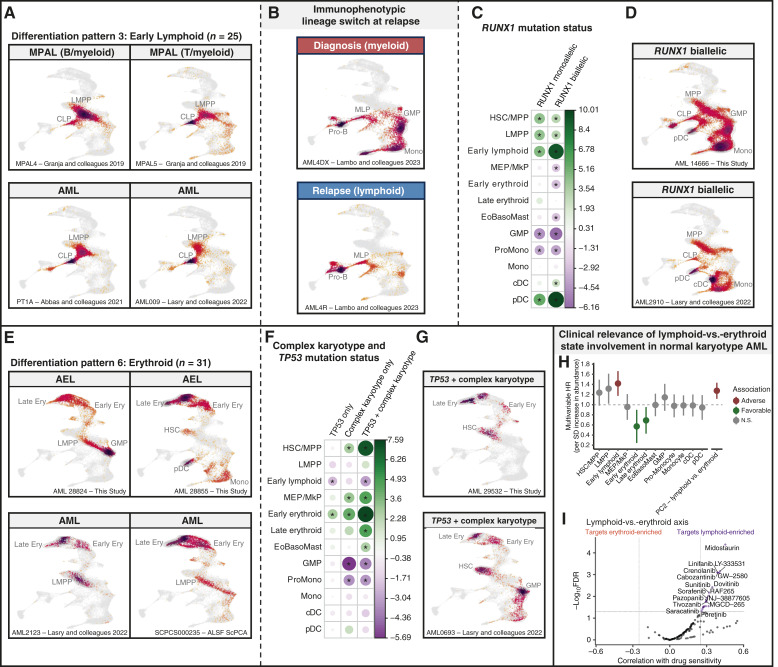
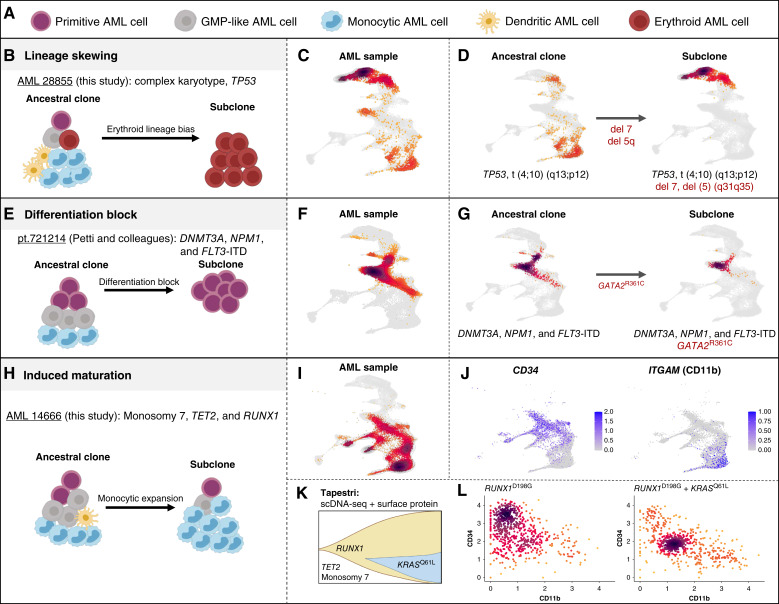
Therapeutic targeting of acute myeloid leukemia (AML) is hampered by intra- and inter-tumoral cell state heterogeneity. To develop a more precise understanding of AML cell states, we constructed a reference atlas of human hematopoiesis from 263,159 single-cell transcriptomes spanning 55 cellular states. Using this atlas, we mapped more than 1.2 million cells spanning 318 leukemia samples, revealing 12 recurrent patterns of aberrant differentiation in AML. Notably, this uncovered unexpected AML cell states resembling lymphoid and erythroid progenitors that were prognostic within the clinically heterogeneous context of normal karyotype AML, independent of genomic classifications. Systematic mapping of genotype-to-phenotype associations revealed specific differentiation landscapes associated with more than 45 genetic drivers. Importantly, distinct cellular hierarchies can arise from samples sharing the same genetic driver, potentially reflecting distinct cellular origins for disease-sustaining leukemia stem cells. Thus, precise mapping of malignant cell states provides insights into leukemogenesis and refines disease classification in acute leukemia.
Significance: We present a single-cell reference atlas of human hematopoiesis and a computational tool for rapid mapping and classification of healthy and leukemic cells. Applied to AML, this has enabled single-cell analysis at the scale of hundreds of patient samples, revealing the full breadth of derailment of differentiation in AML. See related commentary by Berger and Penter, p. 280.
©2025 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
I. Iacobucci reports other support from Arima Genomics, Mission Bio, and Takara outside the submitted work. J.A. Kennedy reports personal fees from Novartis outside the submitted work. C.G. Mullighan reports grants from the NCI and Alex’s Lemonade Stand Foundation for Childhood Cancer during the conduct of the study, as well as personal fees from Illumima and Amgen and grants from Pfizer outside the submitted work. J.E. Dick reports grants from Celgene/Bristol Myers Squibb and other support from Trillium Therapeutics/Pfizer outside the submitted work. No disclosures were reported by the other authors.
Figures

Update of
-
Single-cell transcriptional mapping reveals genetic and non-genetic determinants of aberrant differentiation in AML.bioRxiv [Preprint]. 2024 Nov 1:2023.12.26.573390. doi: 10.1101/2023.12.26.573390. bioRxiv. 2024. Update in: Blood Cancer Discov. 2025 Jul 1;6(4):307-324. doi: 10.1158/2643-3230.BCD-24-0342. PMID: 38234771 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
- RN380110-409786/Canadian Institutes of Health Research (CIHR)
- 1106/Terry Fox Foundation (La Fondation Terry Fox)
- Government of Ontario (Ontario Government)
- American Lebanese Syrian Associated Charities (ALSAC)
- International Development Research Centre (IDRC)
- P30 CA021765/CA/NCI NIH HHS/United States
- University of Toronto's Medicine by Design initiative
- 703212/Canadian Cancer Society (CCS)
- Alex's Lemonade Stand Foundation for Childhood Cancer (ALSF)
- R35 CA197695/CA/NCI NIH HHS/United States
- Henry Schueler 41&9 Foundation
- University of Toronto MD/PhD Studentship Award
- Princess Margaret Cancer Foundation (PMCF)
- Canada Research Chairs (Chaires de recherche du Canada)
- Canada First Research Excellence Fund (CFREF)
- St. Baldrick's Foundation Robert J. Arceci Innovation Award
LinkOut - more resources
Full Text Sources
Medical
