

Comprehensive comparison of the third-generation sequencing tools for bacterial 6mA profiling
- PMID: 40295502
- PMCID: PMC12037826
- DOI: 10.1038/s41467-025-59187-2
Comprehensive comparison of the third-generation sequencing tools for bacterial 6mA profiling
Abstract
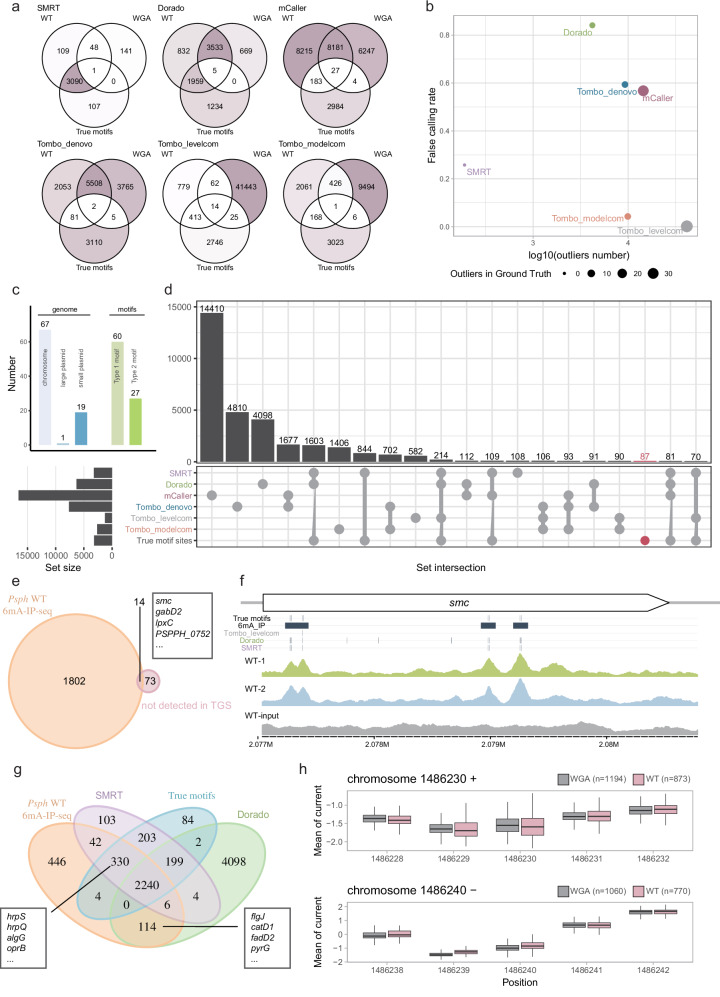
DNA N6-methyladenine (6mA) serves as an intrinsic and principal epigenetic marker in prokaryotes, impacting various biological processes. To date, limited advanced sequencing technologies and analyzing tools are available for bacterial DNA 6mA. Here, we evaluate eight tools designed for the 6mA identification or de novo methylation detection. This assessment includes Nanopore (R9 and R10), Single-Molecule Real-Time (SMRT) Sequencing, and cross-reference with 6mA-IP-seq and DR-6mA-seq. Our multi-dimensional evaluation report encompasses motif discovery, site-level accuracy, single-molecule accuracy, and outlier detection across six bacteria strains. While most tools correctly identify motifs, their performance varies at single-base resolution, with SMRT and Dorado consistently delivering strong performance. Our study indicates that existing tools cannot accurately detect low-abundance methylation sites. Additionally, we introduce an optimized method for advancing 6mA prediction, which substantially improves the detection performance of Dorado. Overall, our study provides a robust and detailed examination of computational tools for bacterial 6mA profiling, highlighting insights for further tool enhancement and epigenetic research.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures





References
-
- Johnson, T. B. & Coghill, R. D. Researches on pyrimidines. C111. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle bacillus. J. Am. Chem. Soc.47, 2838–2844 (1925). - DOI
-
- Waddington C. H. The epigenotype. Endeavour1, 18–20 (1942).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
