

This is a preprint.
Saturation genome editing of RNU4-2 reveals distinct dominant and recessive neurodevelopmental disorders
- PMID: 40297424
- PMCID: PMC12036422
- DOI: 10.1101/2025.04.08.25325442
Saturation genome editing of RNU4-2 reveals distinct dominant and recessive neurodevelopmental disorders
Abstract
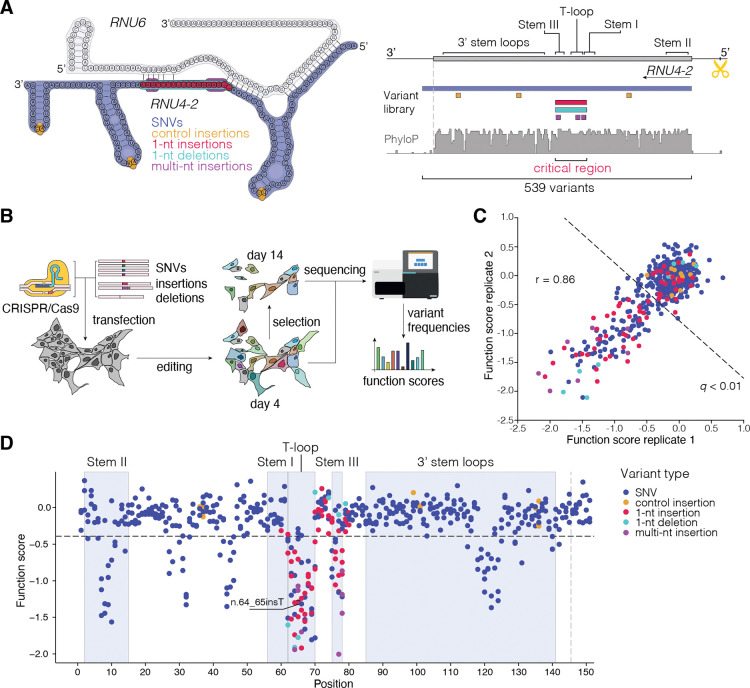
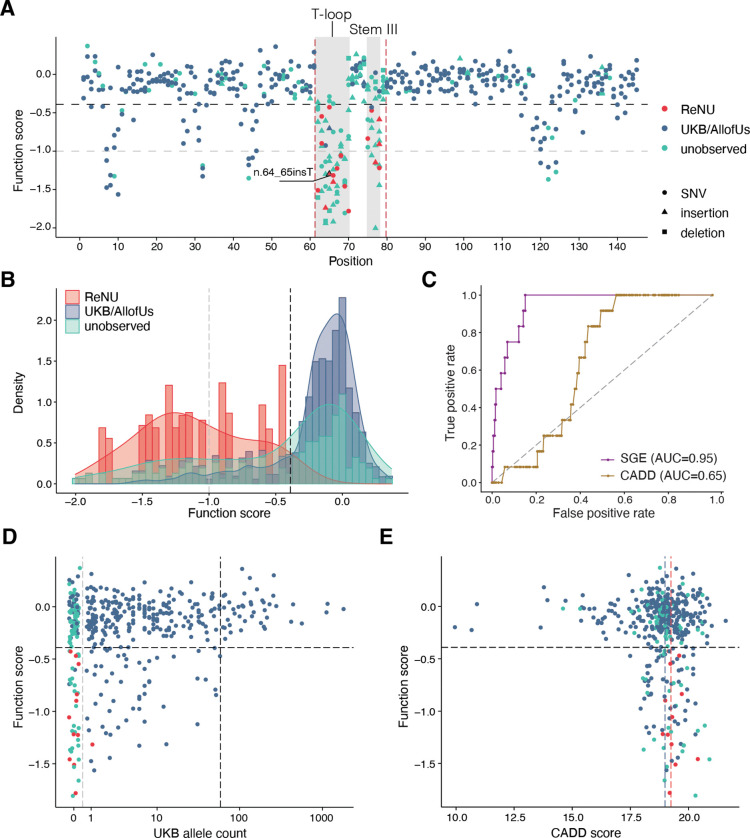
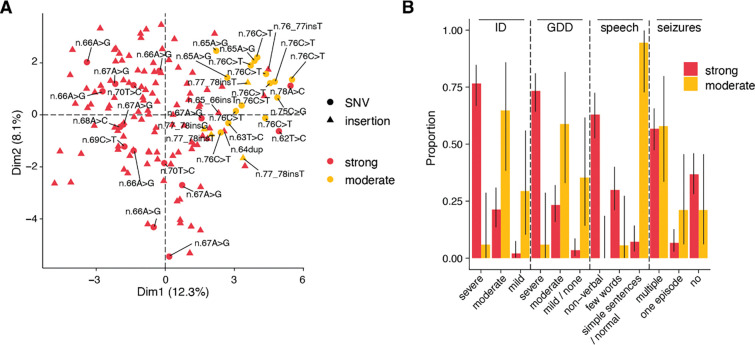
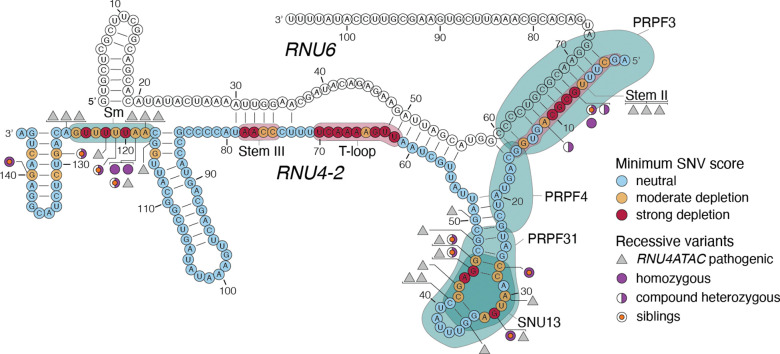
Recently, de novo variants in an 18 nucleotide region in the centre of RNU4-2 were shown to cause ReNU syndrome, a syndromic neurodevelopmental disorder (NDD) that is predicted to affect tens of thousands of individuals worldwide1,2. RNU4-2 is a non-protein-coding gene that is transcribed into the U4 small nuclear RNA (snRNA) component of the major spliceosome3. ReNU syndrome variants disrupt spliceosome function and alter 5' splice site selection1,4. Here, we performed saturation genome editing (SGE) of RNU4-2 to identify the functional and clinical impact of variants across the entire gene. The resulting SGE function scores, derived from variants' effects on cell fitness, discriminate ReNU syndrome variants from those observed in the population and dramatically outperform in silico variant effect prediction. Using these data, we redefine the ReNU syndrome critical region at single nucleotide resolution, resolve variant pathogenicity for variants of uncertain significance, and show that SGE function scores delineate variants by phenotypic severity. Further, we identify variants impacting function in regions of RNU4-2 that are critical for interactions with other spliceosome components. We show that these variants cause a novel recessive NDD that is clinically distinct from ReNU syndrome. Together, this work defines the landscape of variant function across RNU4-2, providing critical insights for both diagnosis and therapeutic development.
Keywords: ReNU syndrome; Saturation genome editing; clinical variant interpretation; neurodevelopmental disorders; non-coding RNA; recessive; small nuclear RNA.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources