

Hydrogen Bonds under Electric Fields with Quantum Accuracy
- PMID: 40298002
- PMCID: PMC12067437
- DOI: 10.1021/acs.jpca.5c01095
Hydrogen Bonds under Electric Fields with Quantum Accuracy
Abstract
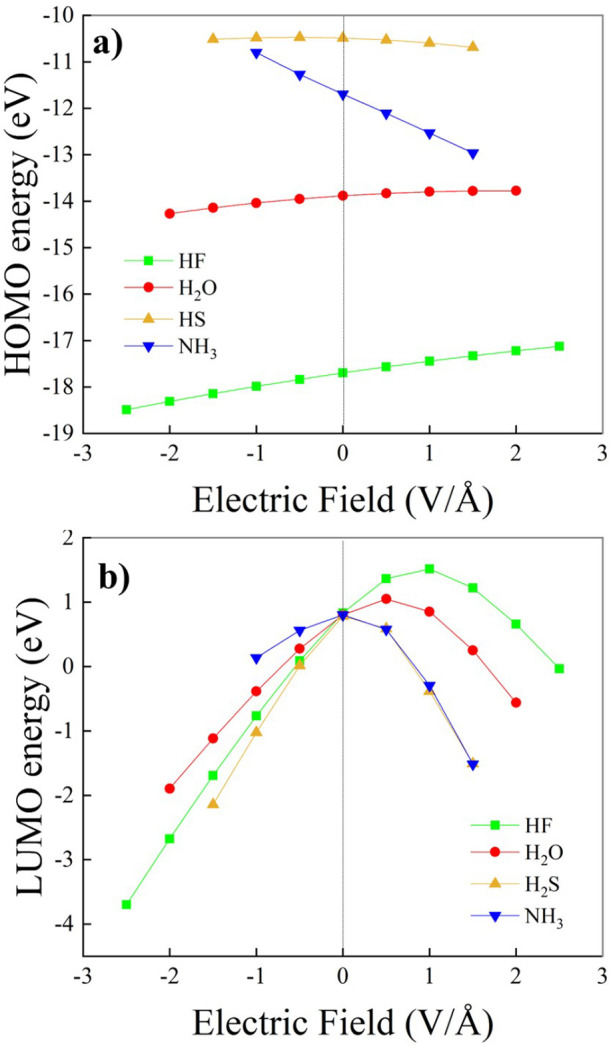
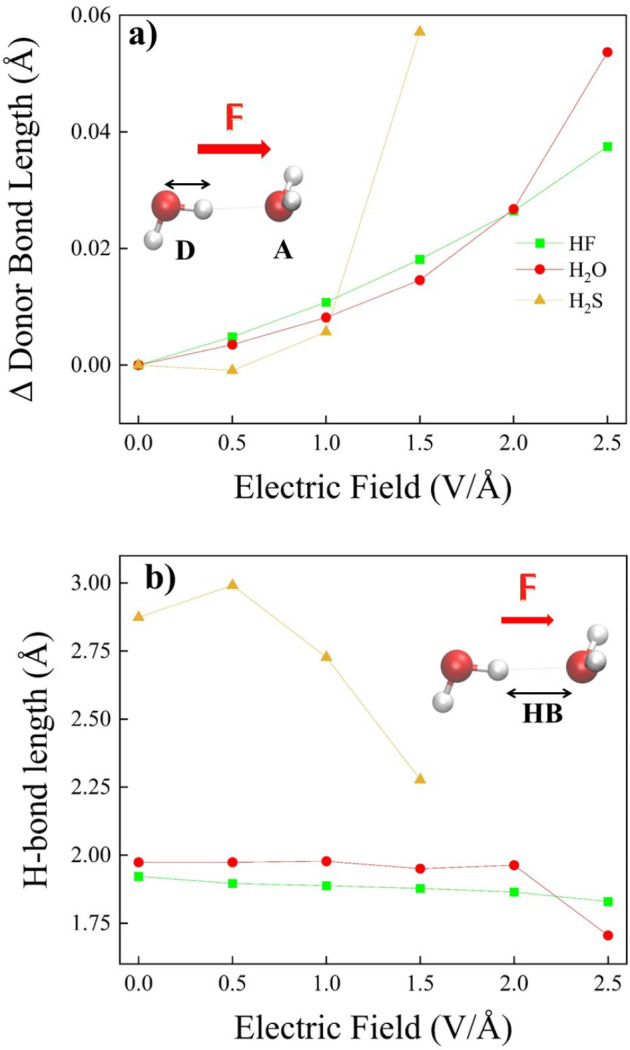
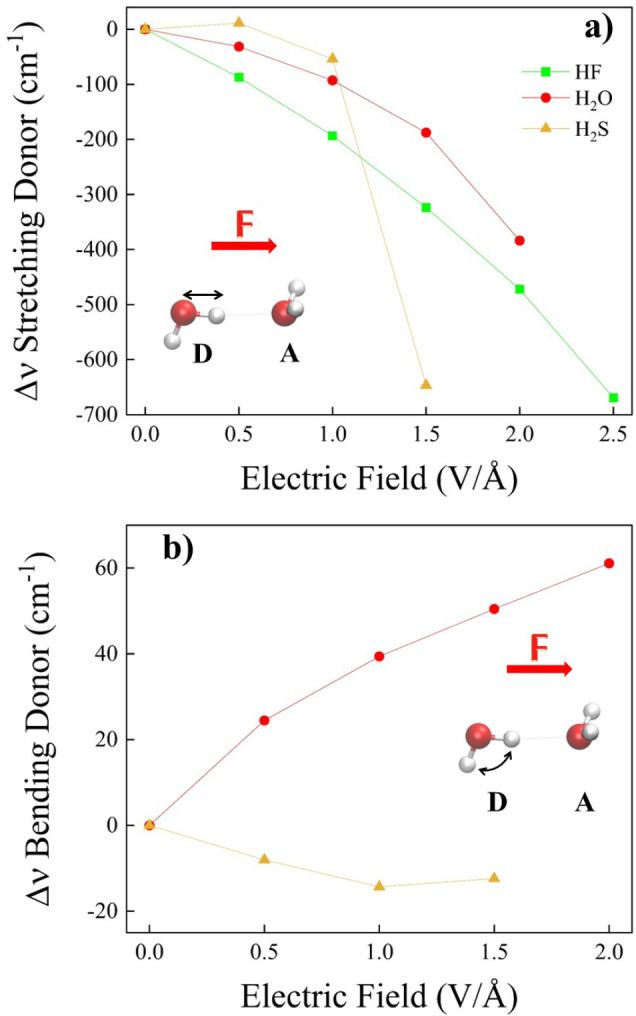
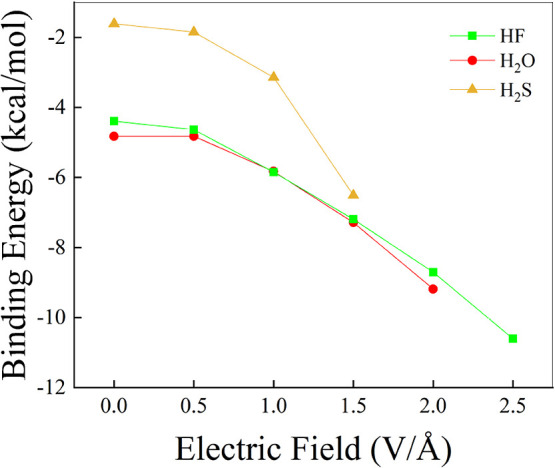
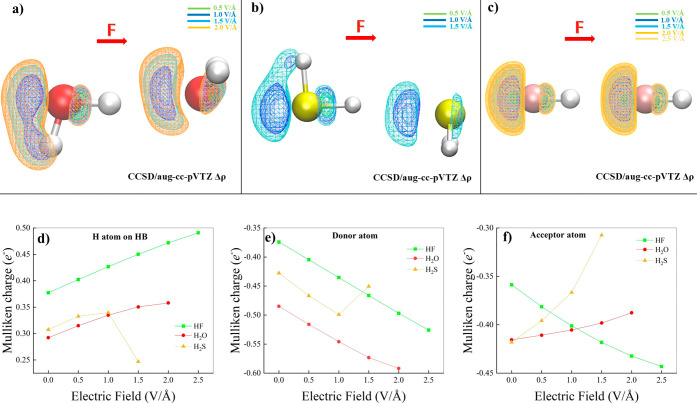
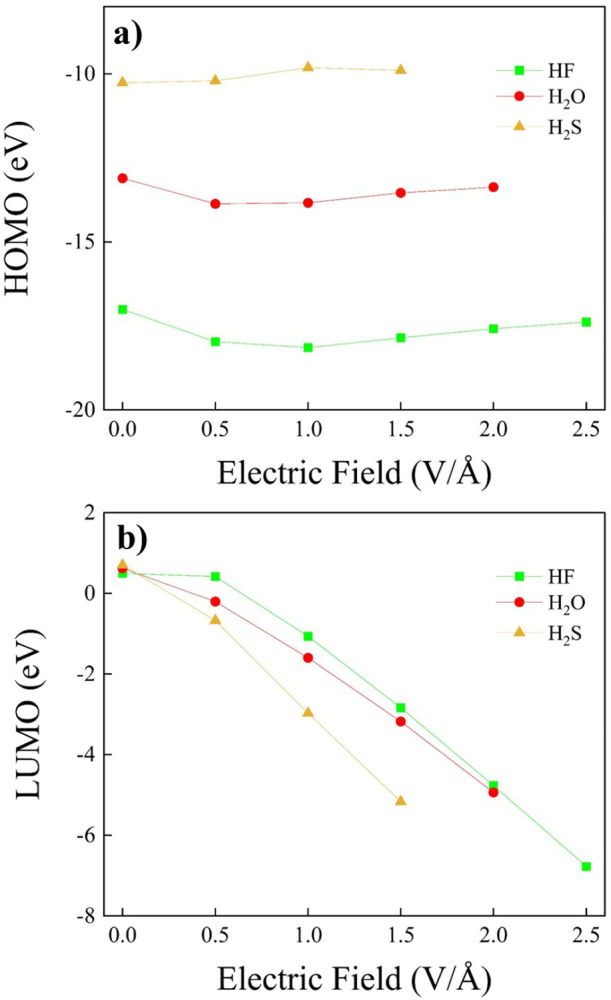
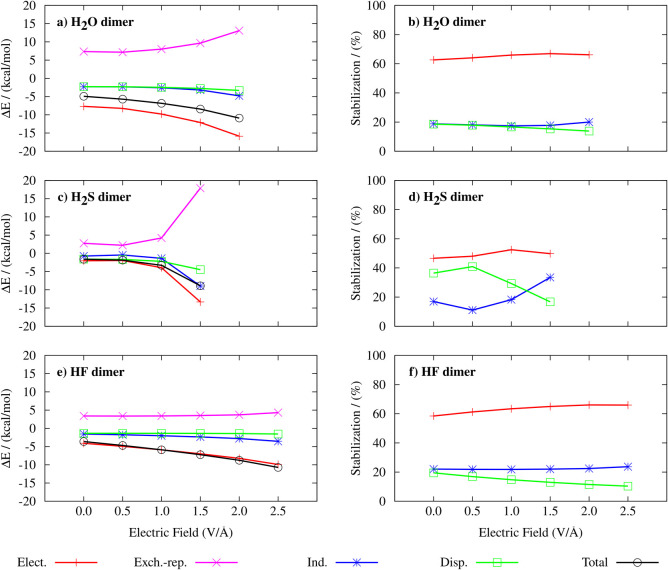
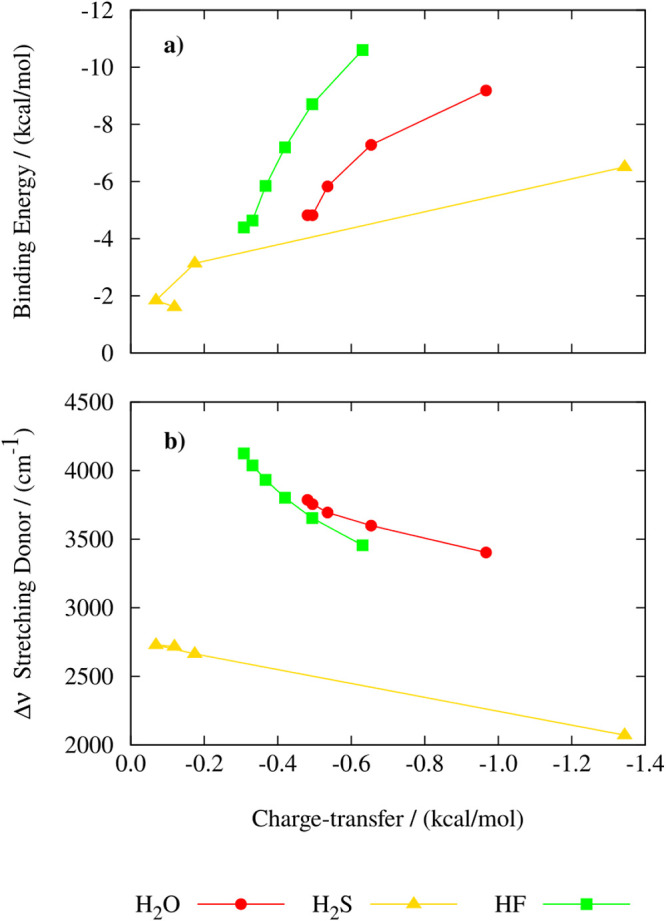
Hydrogen bonds (H-bonds) are pivotal in various chemical and biological systems and exhibit complex behavior under external perturbations. This study investigates the structural, vibrational, and energetic properties of prototypical H-bonded dimers, water (H2O)2, hydrogen fluoride (HF)2, hydrogen sulfide (H2S)2, and ammonia (NH3)2 - and the respective monomers under static and homogeneous electric fields (EFs) using the accurate explicitly correlated singles and doubles coupled cluster method (CCSD) for equilibrium geometries and harmonic vibrational frequencies and the perturbative triples CCSD(T) method for energies. As for the vibrational response of the H2O, HF, H2S, and NH3 monomers, it turns out that dipole derivatives primarily govern the geometry relaxation. Perturbation theory including cubic anharmonicity can reproduce CCSD results on the vibrational Stark effect, except for NH3, where deviations arise due to its floppiness. The field-induced modifications in H-bond lengths, vibrational Stark effects, binding energies, and charge-transfer mechanisms in monomers and dimers are elucidated. Symmetry-adapted perturbation theory (SAPT) analysis on dimers reveals that electrostatics dominates the stabilization of H-bonds across all field strengths, while induction contributions increase significantly with stronger fields, particularly in systems with more polarizable atoms. Our results reveal a universal strengthening of intermolecular interactions at moderate to strong field intensities with significant variability among dimers due to inherent differences in molecular polarizability and charge distribution. Notably, a direct correlation is observed between the binding energies and the vibrational Stark effect of the stretching mode of the H-bond donor molecule, both in relation to the charge-transfer energy term, across all of the investigated dimers. All of these findings provide insights into the EF-driven modulation of H-bonds, highlighting implications for catalysis, hydrogen-based technologies, and biological processes.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
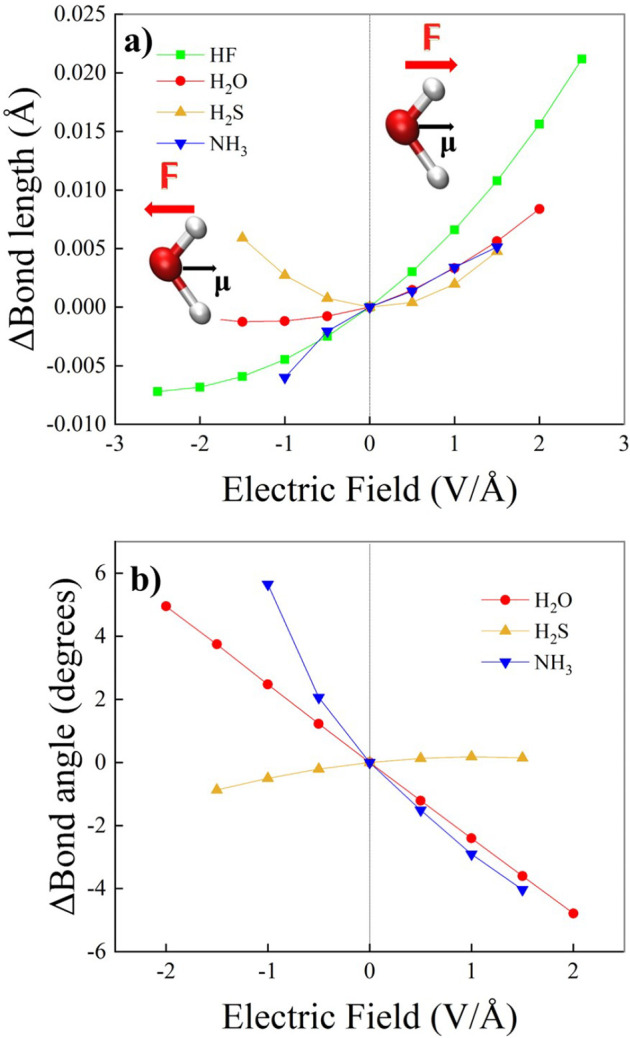
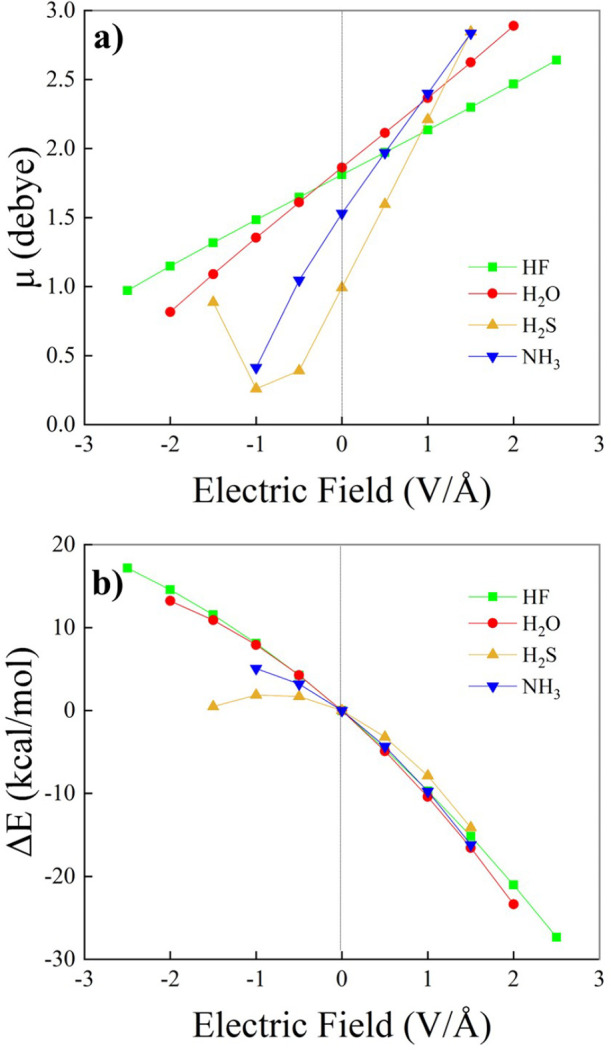
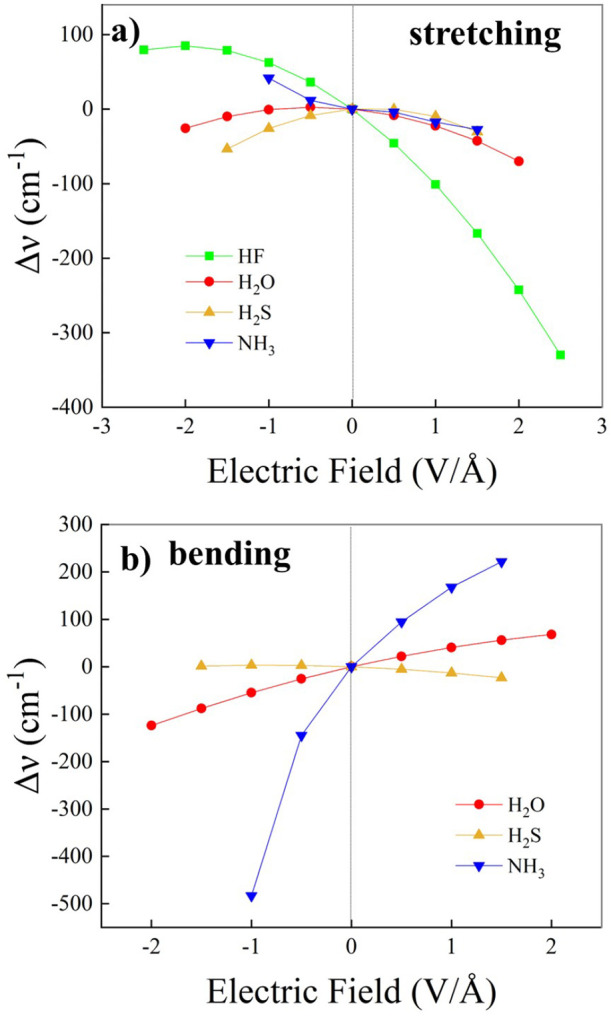
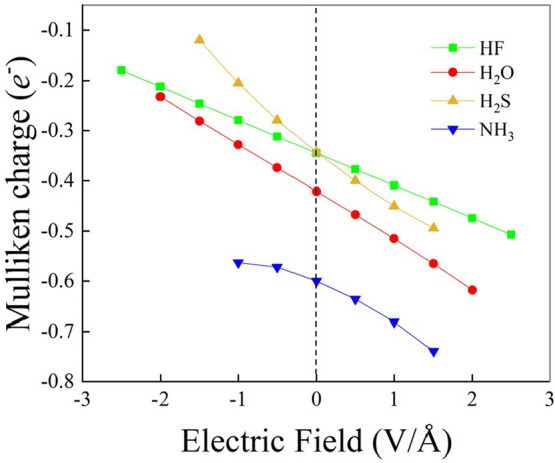
References
-
- Arunan E.; Desiraju G. R.; Klein R. A.; Sadlej J.; Scheiner S.; Alkorta I.; Clary D. C.; Crabtree R. H.; Dannenberg J. J.; et al. Definition of the hydrogen bond (iupac recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. 10.1351/PAC-REC-10-01-02. - DOI
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
