

Major Oxidative and Antioxidant Mechanisms During Heat Stress-Induced Oxidative Stress in Chickens
- PMID: 40298812
- PMCID: PMC12023971
- DOI: 10.3390/antiox14040471
Major Oxidative and Antioxidant Mechanisms During Heat Stress-Induced Oxidative Stress in Chickens
Abstract
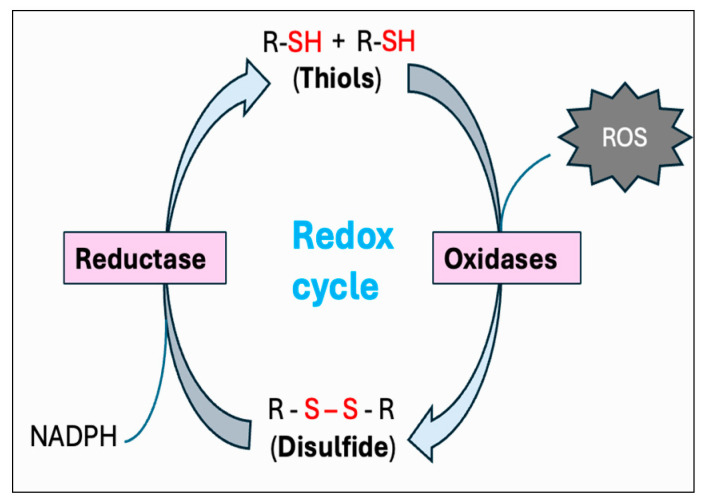
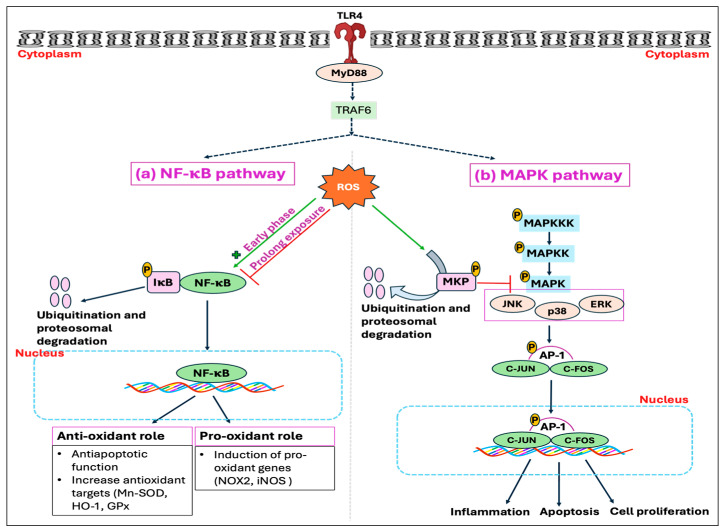
Heat stress (HS) is one of the most important stressors in chickens, and its adverse effects are primarily caused by disturbing the redox homeostasis. An increase in electron leakage from the mitochondrial electron transport chain is the major source of free radical production under HS, which triggers other enzymatic systems to generate more radicals. As a defense mechanism, cells have enzymatic and non-enzymatic antioxidant systems that work cooperatively against free radicals. The generation of free radicals, particularly the reactive oxygen species (ROS) and reactive nitrogen species (RNS), under HS condition outweighs the cellular antioxidant capacity, resulting in oxidative damage to macromolecules, including lipids, carbohydrates, proteins, and DNA. Understanding these detrimental oxidative processes and protective defense mechanisms is important in developing mitigation strategies against HS. This review summarizes the current understanding of major oxidative and antioxidant systems and their molecular mechanisms in generating or neutralizing the ROS/RNS. Importantly, this review explores the potential mechanisms that lead to the development of oxidative stress in heat-stressed chickens, highlighting their unique behavioral and physiological responses against thermal stress. Further, we summarize the major findings associated with these oxidative and antioxidant mechanisms in chickens.
Keywords: antioxidant mechanisms; chickens; heat stress; oxidative stress; reactive oxygen species.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures











Similar articles
-
Redox- and non-redox-metal-induced formation of free radicals and their role in human disease.Arch Toxicol. 2016 Jan;90(1):1-37. doi: 10.1007/s00204-015-1579-5. Epub 2015 Sep 7. Arch Toxicol. 2016. PMID: 26343967 Review.
-
Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia.J Neurol Sci. 2005 Jun 15;233(1-2):145-62. doi: 10.1016/j.jns.2005.03.012. J Neurol Sci. 2005. PMID: 15896810 Review.
-
Free radicals, metals and antioxidants in oxidative stress-induced cancer.Chem Biol Interact. 2006 Mar 10;160(1):1-40. doi: 10.1016/j.cbi.2005.12.009. Epub 2006 Jan 23. Chem Biol Interact. 2006. PMID: 16430879 Review.
-
Association between heat stress and oxidative stress in poultry; mitochondrial dysfunction and dietary interventions with phytochemicals.J Anim Sci Biotechnol. 2016 Jun 28;7:37. doi: 10.1186/s40104-016-0097-5. eCollection 2016. J Anim Sci Biotechnol. 2016. PMID: 27354915 Free PMC article. Review.
-
Effect of heat stress on oxidative damage and antioxidant defense system in white clover (Trifolium repens L.).Planta. 2021 Oct 21;254(5):103. doi: 10.1007/s00425-021-03751-9. Planta. 2021. PMID: 34674051
Cited by
-
Dietary Carnosic Acid Supplementation Improves the Growth Performance, the Antioxidant Status, and Diversity of Intestinal Microbiota in Broilers.Antioxidants (Basel). 2025 Aug 21;14(8):1026. doi: 10.3390/antiox14081026. Antioxidants (Basel). 2025. PMID: 40867922 Free PMC article.
References
-
- Sisein E.A. Biochemistry of free radicals and antioxidants. Sch. Acad. J. Biosci. 2014;2:110–118.
-
- Martemucci G., Costagliola C., Mariano M., D’andrea L., Napolitano P., D’Alessandro A.G. Free radical properties, source and targets, antioxidant consumption and health. Oxygen. 2022;2:48–78. doi: 10.3390/oxygen2020006. - DOI
-
- Halliwell B. eLS. Wiley; Hoboken, NJ, USA: 2001. Free radicals and other reactive species in disease.
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
