

Ivosidenib Confers BRCAness Phenotype and Synthetic Lethality to Poly (ADP-Ribose) Polymerase Inhibition in BRCA1/2-Proficient Cancer Cells
- PMID: 40299557
- PMCID: PMC12025137
- DOI: 10.3390/biomedicines13040958
Ivosidenib Confers BRCAness Phenotype and Synthetic Lethality to Poly (ADP-Ribose) Polymerase Inhibition in BRCA1/2-Proficient Cancer Cells
Abstract
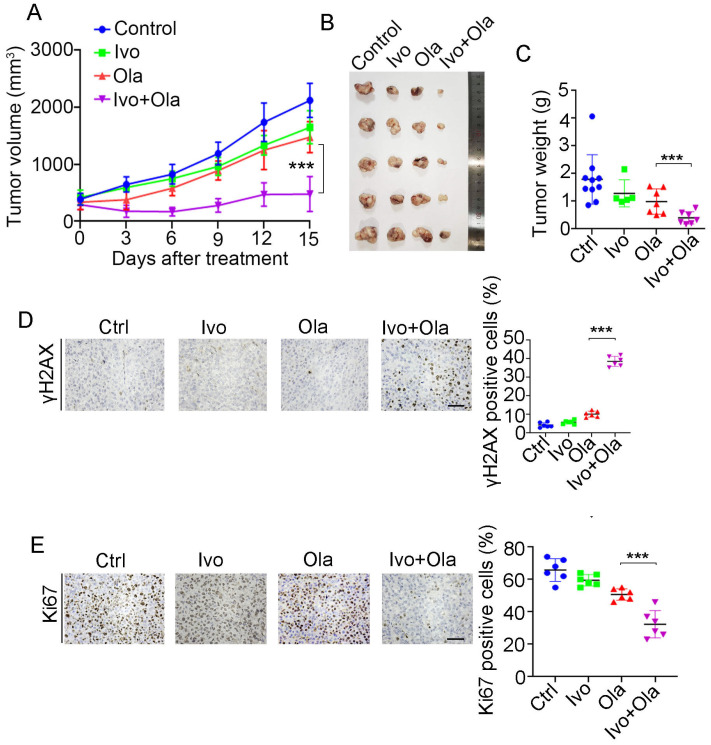
Background/Objectives: PARP inhibitors (PARPi) are pivotal to treating homologous recombination repair-deficient (HRD) cancers, particularly BRCA1/2-mutated ovarian and breast cancers. However, most ovarian and breast cancers harbor wild-type (WT) BRCA1/2, limiting PARPi eligibility. This study aims to identify an approved drug that could induce a BRCAness phenotype, thereby sensitizing WT BRCA cancers to PARPi. Methods: Ovarian and breast cancer cell lines with WT BRCA1/2 were treated with ivosidenib. HR repair efficiency was assessed via RAD51 foci formation and reporter assays. Synthetic lethality with PARPi was evaluated using viability and colony formation assays. Mechanistic studies included RNA-binding protein pulldown, co-immunoprecipitation, and functional analyses of DNA repair pathways. YTHDC2's role in HR was investigated through siRNA knockdown and rescue experiments. Results: Ivosidenib significantly reduced HR repair efficiency and sensitized cells to PARPi, inducing synthetic lethality. Mechanistically, ivosidenib directly bound YTHDC2, an m6A reader critical for HR. This interaction disrupted YTHDC2's ability to promote DNA double-strand break repair via HR, evidenced by impaired recruitment of repair proteins (e.g., BRCA1, RAD51) and accumulation of DNA damage (γH2AX foci). YTHDC2 knockdown phenocopied ivosidenib effects, while overexpression rescued HR defects. Conclusions: Ivosidenib induces BRCAness in WT BRCA ovarian and breast cancers by targeting YTHDC2, thereby suppressing HR repair and enhancing PARPi sensitivity. This uncovers a novel, metabolism-independent mechanism of ivosidenib, repositioning it as a therapeutic agent for HRD tumors. These findings propose a strategy to expand PARPi eligibility to WT BRCA cancers, addressing a critical unmet need in oncology.
Keywords: BRCA1/2; HR repair; Ivosidenib; PARP inhibitors; ovarian cancer.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures






Similar articles
-
Tousled-like kinase loss confers PARP inhibitor resistance in BRCA1-mutated cancers by impeding non-homologous end joining repair.Mol Med. 2025 Jan 22;31(1):18. doi: 10.1186/s10020-025-01066-z. Mol Med. 2025. PMID: 39844055 Free PMC article.
-
Natural product Pulsatilla saponin D sensitizes BRCA-proficient ovarian cancers to PARP inhibitors through inhibiting homologous recombination repair.J Pharm Pharmacol. 2025 Apr 3;77(4):511-523. doi: 10.1093/jpp/rgaf006. J Pharm Pharmacol. 2025. PMID: 40036611
-
Pharmacologic Induction of BRCAness in BRCA-Proficient Cancers: Expanding PARP Inhibitor Use.Cancers (Basel). 2022 May 26;14(11):2640. doi: 10.3390/cancers14112640. Cancers (Basel). 2022. PMID: 35681619 Free PMC article. Review.
-
RAD51-Mediated DNA Homologous Recombination Is Independent of PTEN Mutational Status.Cancers (Basel). 2020 Oct 29;12(11):3178. doi: 10.3390/cancers12113178. Cancers (Basel). 2020. PMID: 33138032 Free PMC article.
-
Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: the rationale for their inclusion in the clinic.J Exp Clin Cancer Res. 2016 Nov 24;35(1):179. doi: 10.1186/s13046-016-0456-2. J Exp Clin Cancer Res. 2016. PMID: 27884198 Free PMC article. Review.
References
-
- Chappidi N., Quail T., Doll S., Vogel L.T., Aleksandrov R., Felekyan S., Kuhnemuth R., Stoynov S., Seidel C.A.M., Brugues J., et al. PARP1-DNA co-condensation drives DNA repair site assembly to prevent disjunction of broken DNA ends. Cell. 2024;187:945–961.e918. doi: 10.1016/j.cell.2024.01.015. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
