

UNISOM: Unified Somatic Calling and Machine Learning-based Classification Enhance the Discovery of CHIP
- PMID: 40300108
- PMCID: PMC12282763
- DOI: 10.1093/gpbjnl/qzaf040
UNISOM: Unified Somatic Calling and Machine Learning-based Classification Enhance the Discovery of CHIP
Abstract
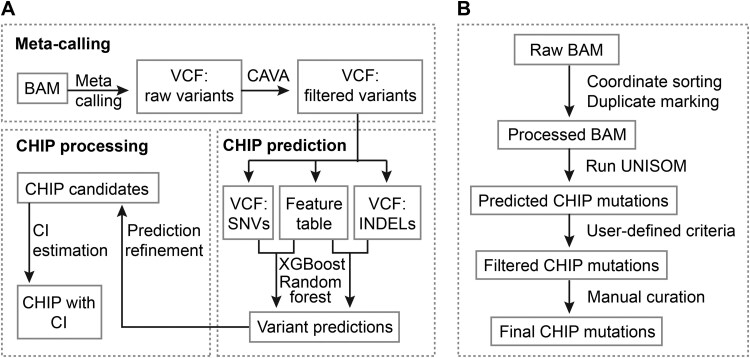
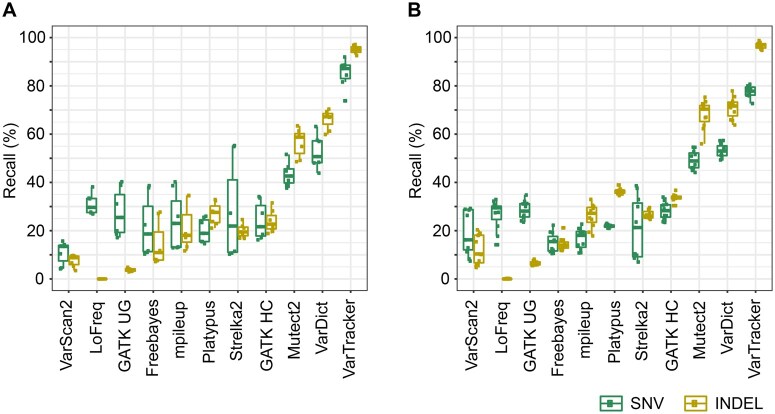
Clonal hematopoiesis (CH) of indeterminate potential (CHIP), driven by somatic mutations in leukemia-associated genes, confers increased risk of hematologic malignancies, cardiovascular disease, and all-cause mortality. In blood of healthy individuals, small CH clones can expand over time to reach 2% variant allele frequency (VAF), the current threshold for CHIP. Nevertheless, reliable detection of low-VAF CHIP mutations is challenging, often relying on deep targeted sequencing. Here, we present UNISOM, a streamlined workflow for enhancing CHIP detection from whole-genome and whole-exome sequencing data that are underpowered, especially for low VAFs. UNISOM utilizes a meta-caller for variant detection, in couple with machine learning models which classify variants into CHIP, germline, and artifact. In whole-exome sequencing data, UNISOM recovered nearly 80% of the CHIP mutations identified via deep targeted sequencing in the same cohort. Applied to whole-genome sequencing data from Mayo Clinic Biobank, it recapitulated the patterns previously established in much larger cohorts, including the most frequently mutated CHIP genes and predominant mutation types and signatures, as well as strong associations of CHIP with age and smoking status. Notably, 30% of the identified CHIP mutations had < 5% VAFs, demonstrating its high sensitivity toward small mutant clones. This workflow is applicable to CHIP screening in population genomic studies. The UNISOM pipeline is freely available at https://github.com/shulanmayo/UNISOM and https://ngdc.cncb.ac.cn/biocode/tool/7816.
Keywords: Clonal hematopoiesis of indeterminate potential; Machine learning; Somatic variant calling; Whole-exome sequencing; Whole-genome sequencing.
© The Author(s) 2025. Published by Oxford University Press and Science Press on behalf of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China.
Conflict of interest statement
The authors have declared no competing interests.
Figures




Similar articles
-
Can a Liquid Biopsy Detect Circulating Tumor DNA With Low-passage Whole-genome Sequencing in Patients With a Sarcoma? A Pilot Evaluation.Clin Orthop Relat Res. 2025 Jan 1;483(1):39-48. doi: 10.1097/CORR.0000000000003161. Epub 2024 Jun 21. Clin Orthop Relat Res. 2025. PMID: 38905450
-
Association of Diet Quality With Prevalence of Clonal Hematopoiesis and Adverse Cardiovascular Events.JAMA Cardiol. 2021 Sep 1;6(9):1069-1077. doi: 10.1001/jamacardio.2021.1678. JAMA Cardiol. 2021. PMID: 34106216 Free PMC article.
-
Exploring potential therapeutic targets for colorectal tumors based on whole genome sequencing of colorectal tumors and paracancerous tissues.Front Mol Biosci. 2025 Jul 4;12:1605117. doi: 10.3389/fmolb.2025.1605117. eCollection 2025. Front Mol Biosci. 2025. PMID: 40688112 Free PMC article.
-
Clonal hematopoiesis of indeterminate potential and cardiovascular disease.Transl Res. 2023 May;255:152-158. doi: 10.1016/j.trsl.2022.08.013. Epub 2022 Sep 5. Transl Res. 2023. PMID: 36067904 Free PMC article. Review.
-
Clonal hematopoiesis of indeterminate potential (CHIP) in cerebromicrovascular aging: implications for vascular contributions to cognitive impairment and dementia (VCID).Geroscience. 2025 Jun;47(3):2739-2775. doi: 10.1007/s11357-025-01654-1. Epub 2025 Apr 11. Geroscience. 2025. PMID: 40214958 Free PMC article. Review.
References
-
- Steensma DP. Clinical implications of clonal hematopoiesis. Mayo Clin Proc 2018;93:1122–30. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
