

Analytic Approaches in Genomic Epidemiological Studies of Parasitic Protozoa
- PMID: 40303014
- PMCID: PMC12017464
- DOI: 10.1155/2024/7679727
Analytic Approaches in Genomic Epidemiological Studies of Parasitic Protozoa
Abstract
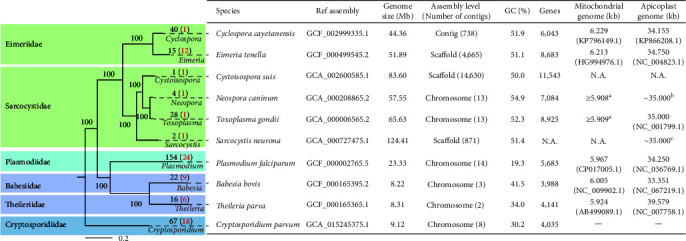
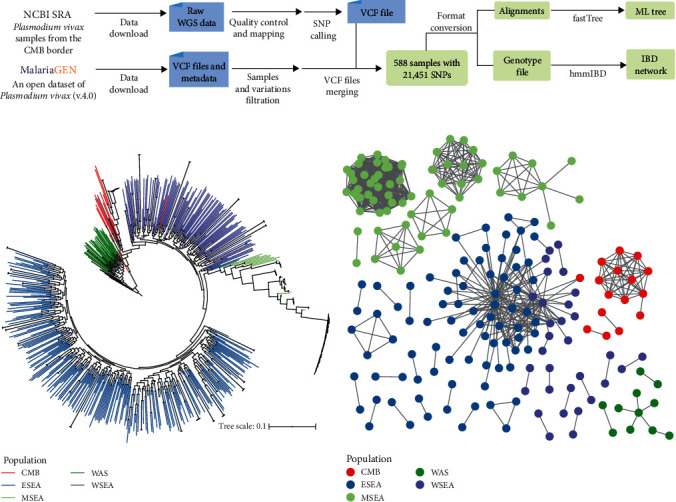
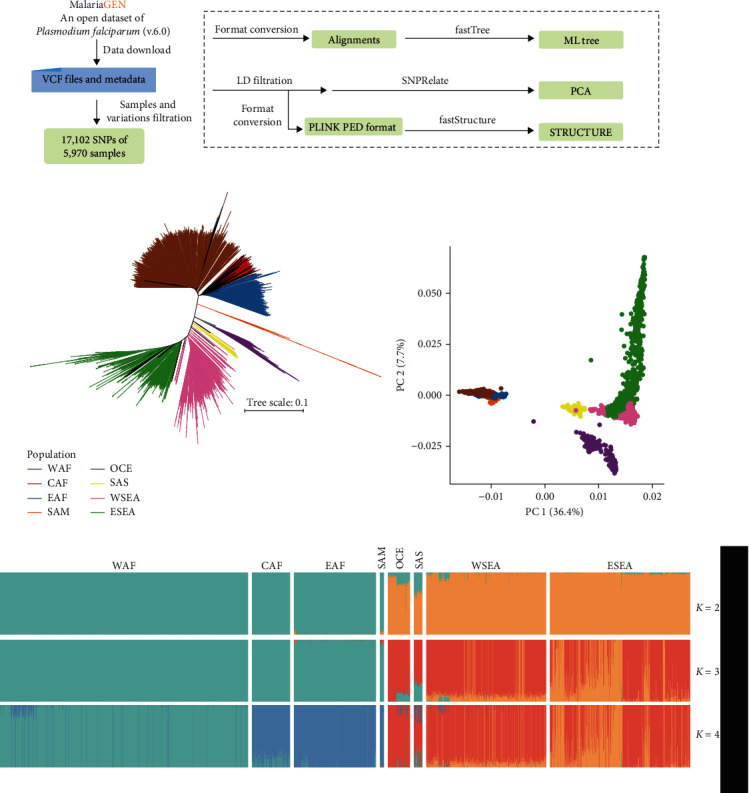
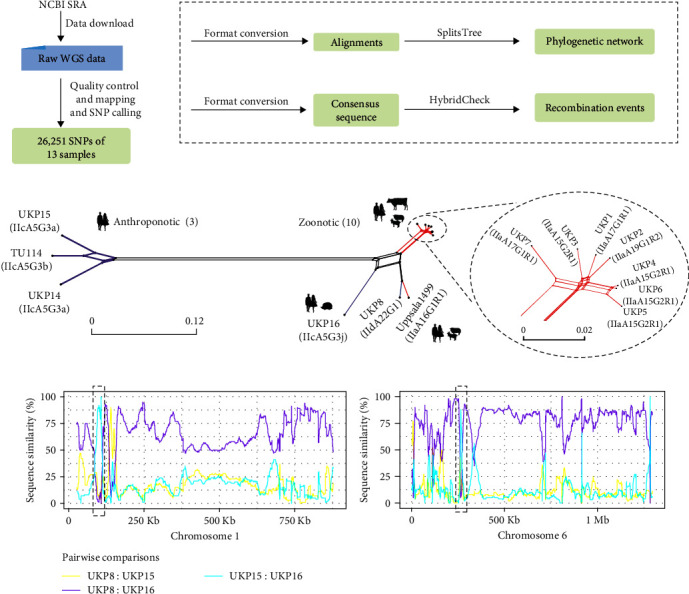
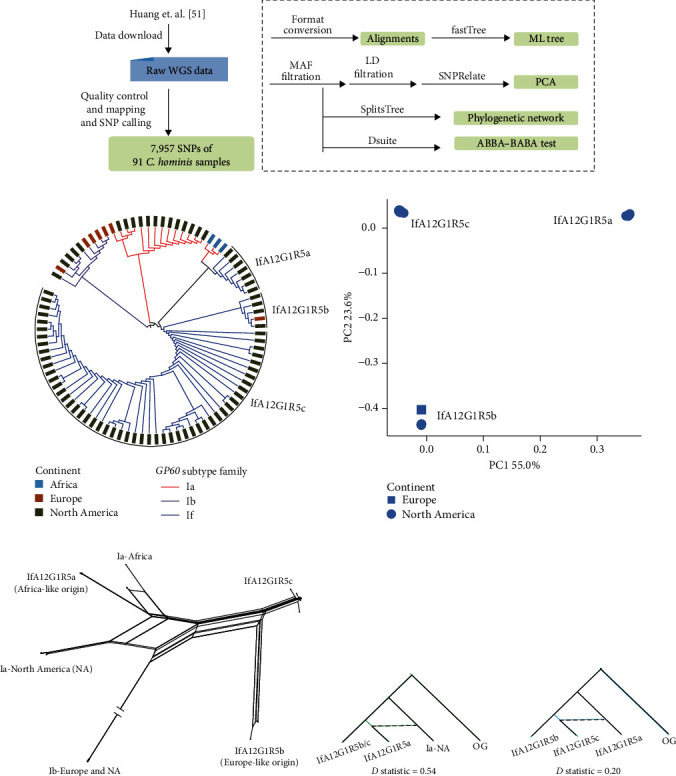
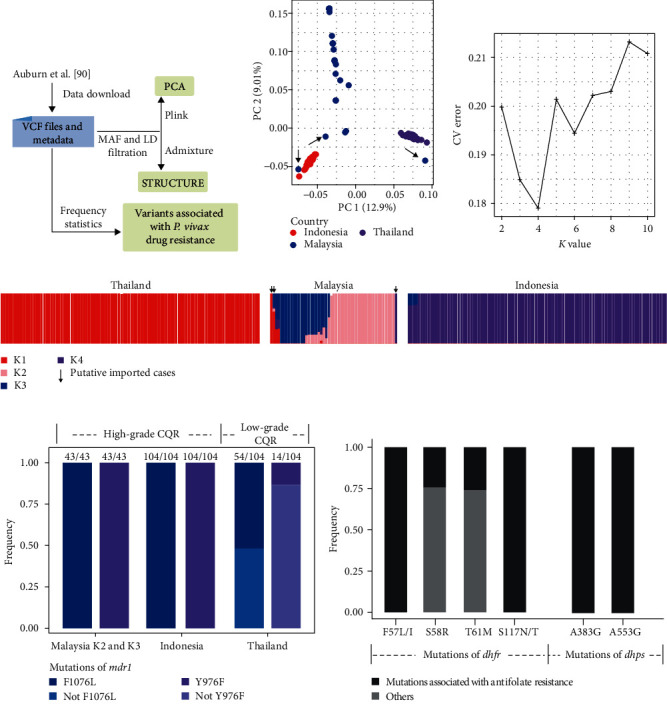
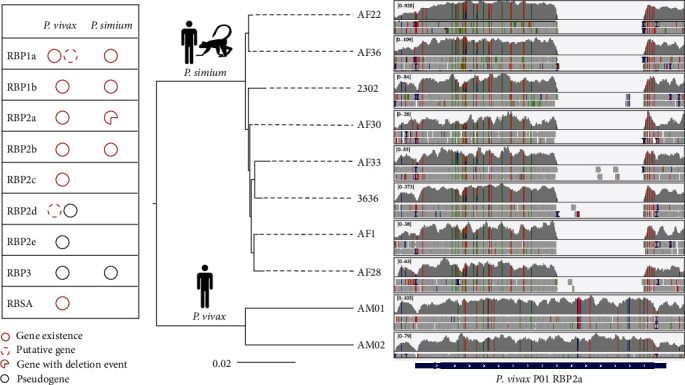
Whole genome sequencing (WGS) plays an important role in the advanced characterization of pathogen transmission and is widely used in studies of major bacterial and viral diseases. Although protozoan parasites cause serious diseases in humans and animals, WGS data on them are relatively scarce due to the large genomes and lack of cultivation techniques for some. In this review, we have illustrated bioinformatic analyses of WGS data and their applications in studies of the genomic epidemiology of apicomplexan parasites. WGS has been used in outbreak detection and investigation, studies of pathogen transmission and evolution, and drug resistance surveillance and tracking. However, comparative analysis of parasite WGS data is still in its infancy, and available WGS data are mainly from a few genera of major public health importance, such as Plasmodium, Toxoplasma, and Cryptosporidium. In addition, the utility of third-generation sequencing technology for complete genome assembly at the chromosome level, studies of the biological significance of structural genomic variation, and molecular surveillance of pathogens has not been fully exploited. These issues require large-scale WGS of various protozoan parasites of public health and veterinary importance using both second- and third-generation sequencing technologies.
Copyright © 2024 Tianpeng Wang et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
Similar articles
-
Advances in the application of genetic manipulation methods to apicomplexan parasites.Int J Parasitol. 2017 Oct;47(12):701-710. doi: 10.1016/j.ijpara.2017.08.002. Epub 2017 Sep 8. Int J Parasitol. 2017. PMID: 28893636 Review.
-
[Biology, epidemiology and diagnostics of pathogenic waterborne protozoan parasites].Wiad Parazytol. 2010;56(2):125-32. Wiad Parazytol. 2010. PMID: 20707296 Polish.
-
Phylogeny and evolution of apicoplasts and apicomplexan parasites.Parasitol Int. 2015 Jun;64(3):254-9. doi: 10.1016/j.parint.2014.10.005. Epub 2014 Oct 14. Parasitol Int. 2015. PMID: 25451217 Review.
-
Babesia gibsoni endemic to Wuhan, China: mitochondrial genome sequencing, annotation, and comparison with apicomplexan parasites.Parasitol Res. 2019 Jan;118(1):235-243. doi: 10.1007/s00436-018-6158-2. Epub 2018 Nov 24. Parasitol Res. 2019. PMID: 30474737
-
Genomics of apicomplexan parasites.Crit Rev Biochem Mol Biol. 2017 Jun;52(3):254-273. doi: 10.1080/10409238.2017.1290043. Epub 2017 Feb 22. Crit Rev Biochem Mol Biol. 2017. PMID: 28276701 Free PMC article. Review.
Cited by
-
The transformation of a Cryptosporidium reference microbiology service to tackle the One Health challenge.Food Waterborne Parasitol. 2025 Jul 1;40:e00274. doi: 10.1016/j.fawpar.2025.e00274. eCollection 2025 Sep. Food Waterborne Parasitol. 2025. PMID: 40688529 Free PMC article. Review.
References
-
- Dallman T. J., Jalava K., Verlander N. Q., et al. Identification of domestic reservoirs and common exposures in an emerging lineage of Shiga toxin-producing Escherichia coli O157: H7 in England: a genomic epidemiological analysis. Lancet Microbe . 2022;3(8):e606–e615. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
