

Major Shift of Influenza A Virus of Swine (IAV-S) by Human-to-Swine Spillover of the 2009 Pandemic Virus in Korea
- PMID: 40303172
- PMCID: PMC12016703
- DOI: 10.1155/2024/6366170
Major Shift of Influenza A Virus of Swine (IAV-S) by Human-to-Swine Spillover of the 2009 Pandemic Virus in Korea
Abstract
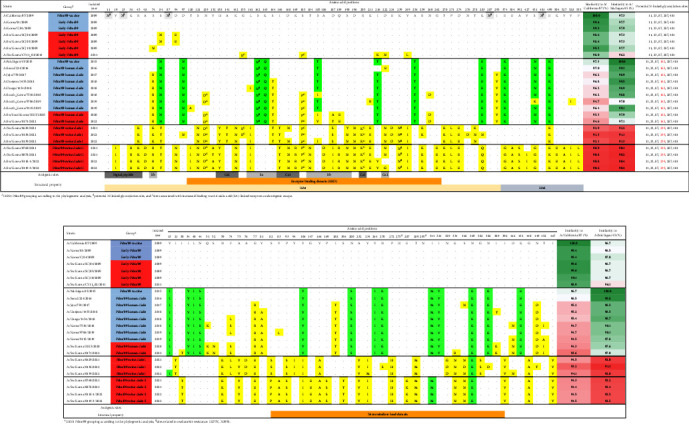
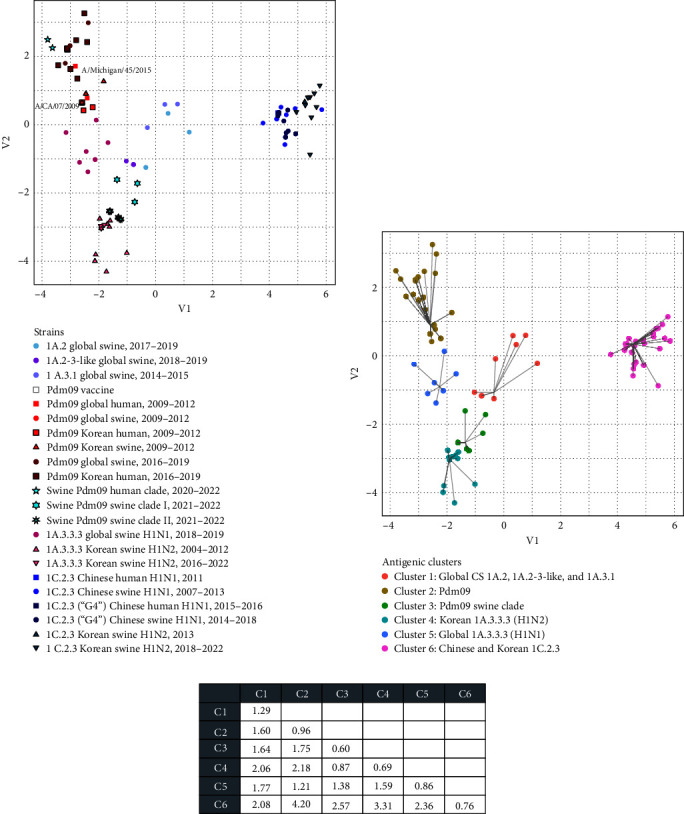
The 2009 influenza A H1N1 pandemic (pdm09) originated from the influenza A virus of swine (IAV-S) through multiple reassortment events with avian and human IAVs. The pdm09 reportedly reintroduced the virus to pigs, contributing to the evolution and diversity of IAV-S through frequent reassortment and drifts. Surveillance and whole-genome sequencing of IAV-S from conventional pig farms in Korea during 2021-2022 revealed that the genetic diversity of H1 and H3 IAV-S was continuously enriched after human-to-swine spillover of pdm09 viruses with long-term maintenance, persistence, and reassortment of virus lineages. Evidence of additional human-to-swine spillover of viruses that are different from the 2009 virus but close to that of the recent H1N1pdm09 human vaccine was identified in this study. The identification of swine-adapted pdm09 viruses, which have accumulated amino acid mutations with potentially altered antigenicity and a unique potential N-glycosylation site within the haemagglutinin (HA) gene, suggests the distinctive evolution of spillover pdm09 viruses in swine. The genetic constellation of the recently emerging Eurasian avian-like swine lineage and the preexisting classical swine lineage H1 viruses in Korea has been expanded through reassortment with cocirculating pdm09 viruses and/or H3N2 IAV-S harboring the pdm09 M gene (H3N2pM). Collectively, after the major shift of Korean IAV-S from the classical swine lineage to the pdm09 lineage in 2009, the frequent spillover of pdm09 viruses and the circulation of IAV-S harboring pdm09 gene segments led to the continuous diversification of IAV-S through antigenic drift and shift, raising concerns about the potential reintroduction of these viruses to humans.
Copyright © 2024 Seung-Chai Kim et al.
Conflict of interest statement
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Figures





Similar articles
-
Influenza A Viruses of Swine (IAV-S) in Vietnam from 2010 to 2015: Multiple Introductions of A(H1N1)pdm09 Viruses into the Pig Population and Diversifying Genetic Constellations of Enzootic IAV-S.J Virol. 2016 Dec 16;91(1):e01490-16. doi: 10.1128/JVI.01490-16. Print 2017 Jan 1. J Virol. 2016. PMID: 27795418 Free PMC article.
-
Reassortment between Swine H3N2 and 2009 Pandemic H1N1 in the United States Resulted in Influenza A Viruses with Diverse Genetic Constellations with Variable Virulence in Pigs.J Virol. 2017 Jan 31;91(4):e01763-16. doi: 10.1128/JVI.01763-16. Print 2017 Feb 15. J Virol. 2017. PMID: 27928015 Free PMC article.
-
Emergence of H3N2pM-like and novel reassortant H3N1 swine viruses possessing segments derived from the A (H1N1)pdm09 influenza virus, Korea.Influenza Other Respir Viruses. 2013 Nov;7(6):1283-91. doi: 10.1111/irv.12154. Epub 2013 Aug 30. Influenza Other Respir Viruses. 2013. PMID: 24034626 Free PMC article.
-
[Swine influenza virus: evolution mechanism and epidemic characterization--a review].Wei Sheng Wu Xue Bao. 2009 Sep;49(9):1138-45. Wei Sheng Wu Xue Bao. 2009. PMID: 20030049 Review. Chinese.
-
Genetics, evolution, and the zoonotic capacity of European Swine influenza viruses.Curr Top Microbiol Immunol. 2013;370:29-55. doi: 10.1007/82_2012_267. Curr Top Microbiol Immunol. 2013. PMID: 23011571 Review.
Cited by
-
Transmission and Pathologic Findings of Divergent Human Seasonal H1N1pdm09 Influenza A Viruses Following Spillover Into Pigs in the United States.Influenza Other Respir Viruses. 2025 Jul;19(7):e70128. doi: 10.1111/irv.70128. Influenza Other Respir Viruses. 2025. PMID: 40671507 Free PMC article.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
