

GōMartini 3: From large conformational changes in proteins to environmental bias corrections
- PMID: 40307210
- PMCID: PMC12043922
- DOI: 10.1038/s41467-025-58719-0
GōMartini 3: From large conformational changes in proteins to environmental bias corrections
Abstract
Coarse-grained modeling has become an important tool to supplement experimental measurements, allowing access to spatio-temporal scales beyond all-atom based approaches. The GōMartini model combines structure- and physics-based coarse-grained approaches, balancing computational efficiency and accurate representation of protein dynamics with the capabilities of studying proteins in different biological environments. This paper introduces an enhanced GōMartini model, which combines a virtual-site implementation of Gō models with Martini 3. The implementation has been extensively tested by the community since the release of the reparametrized version of Martini. This work demonstrates the capabilities of the model in diverse case studies, ranging from protein-membrane binding to protein-ligand interactions and AFM force profile calculations. The model is also versatile, as it can address recent inaccuracies reported in the Martini protein model. Lastly, the paper discusses the advantages, limitations, and future perspectives of the Martini 3 protein model and its combination with Gō models.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures

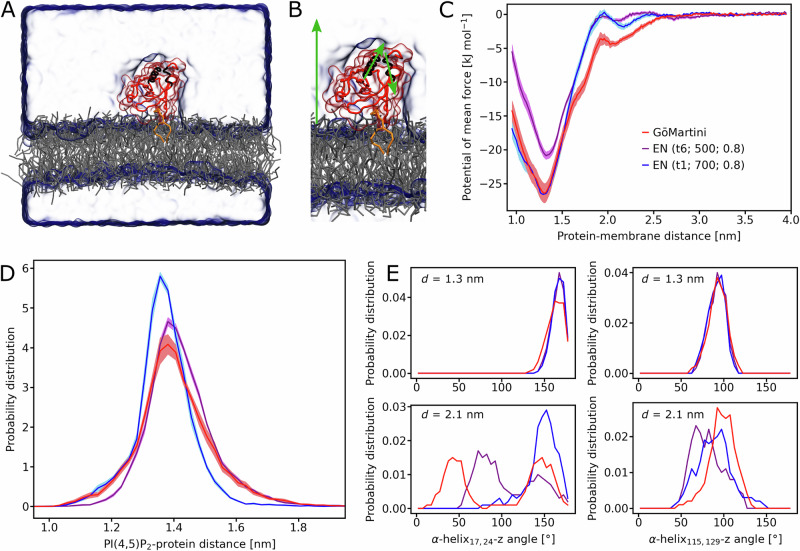
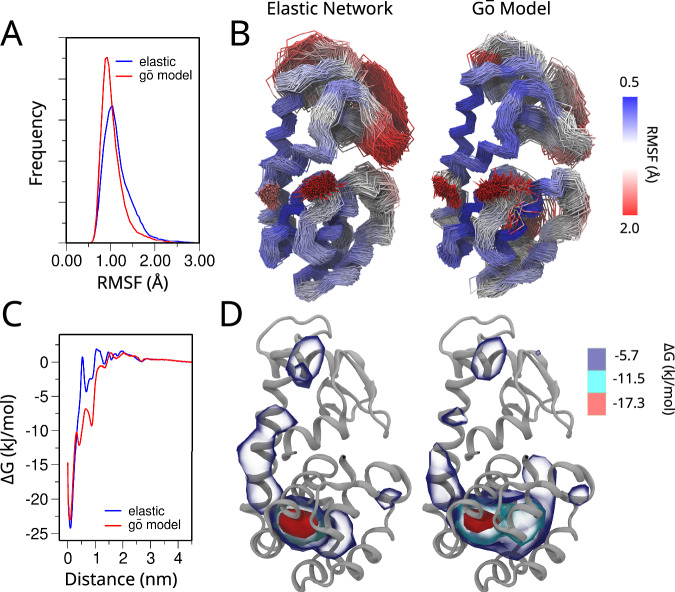

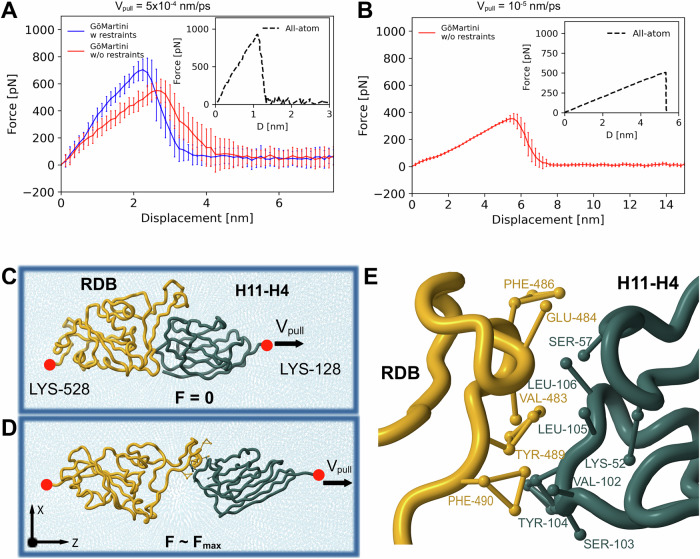
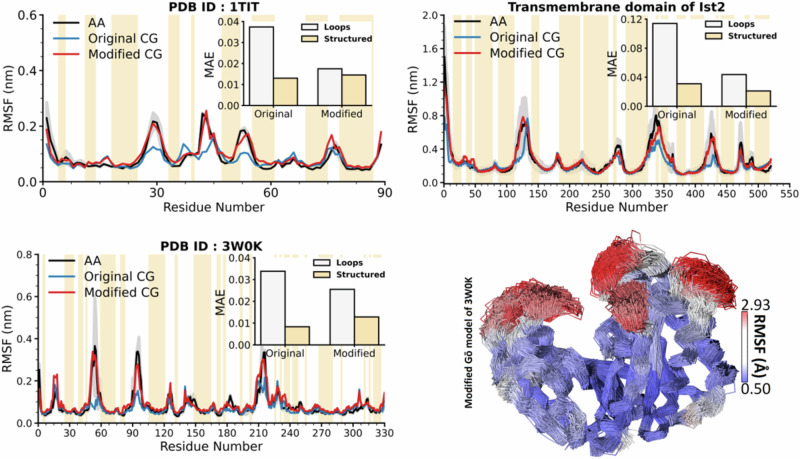

References
-
- Grimaldo, M., Roosen-Runge, F., Zhang, F., Schreiber, F. & Seydel, T. Dynamics of proteins in solution. Q. Rev. Biophys. 52, 10.1017/S0033583519000027 (2019).
MeSH terms
Substances
Grants and funding
- 748895/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 Marie Skłodowska-Curie Actions (H2020 Excellent Science - Marie Skłodowska-Curie Actions)
- CMMS/Hessisches Ministerium für Wissenschaft und Kunst (Hessen State Ministry of Higher Education, Research and the Arts)
- CEECIND/04124/2017/CP1428/CT0008/Ministry of Education and Science | Fundação para a Ciência e a Tecnologia (Portuguese Science and Technology Foundation)
LinkOut - more resources
Full Text Sources
Miscellaneous
