

Selective inhibition of stromal mechanosensing suppresses cardiac fibrosis
- PMID: 40307543
- PMCID: PMC12176515
- DOI: 10.1038/s41586-025-08945-9
Selective inhibition of stromal mechanosensing suppresses cardiac fibrosis
Abstract
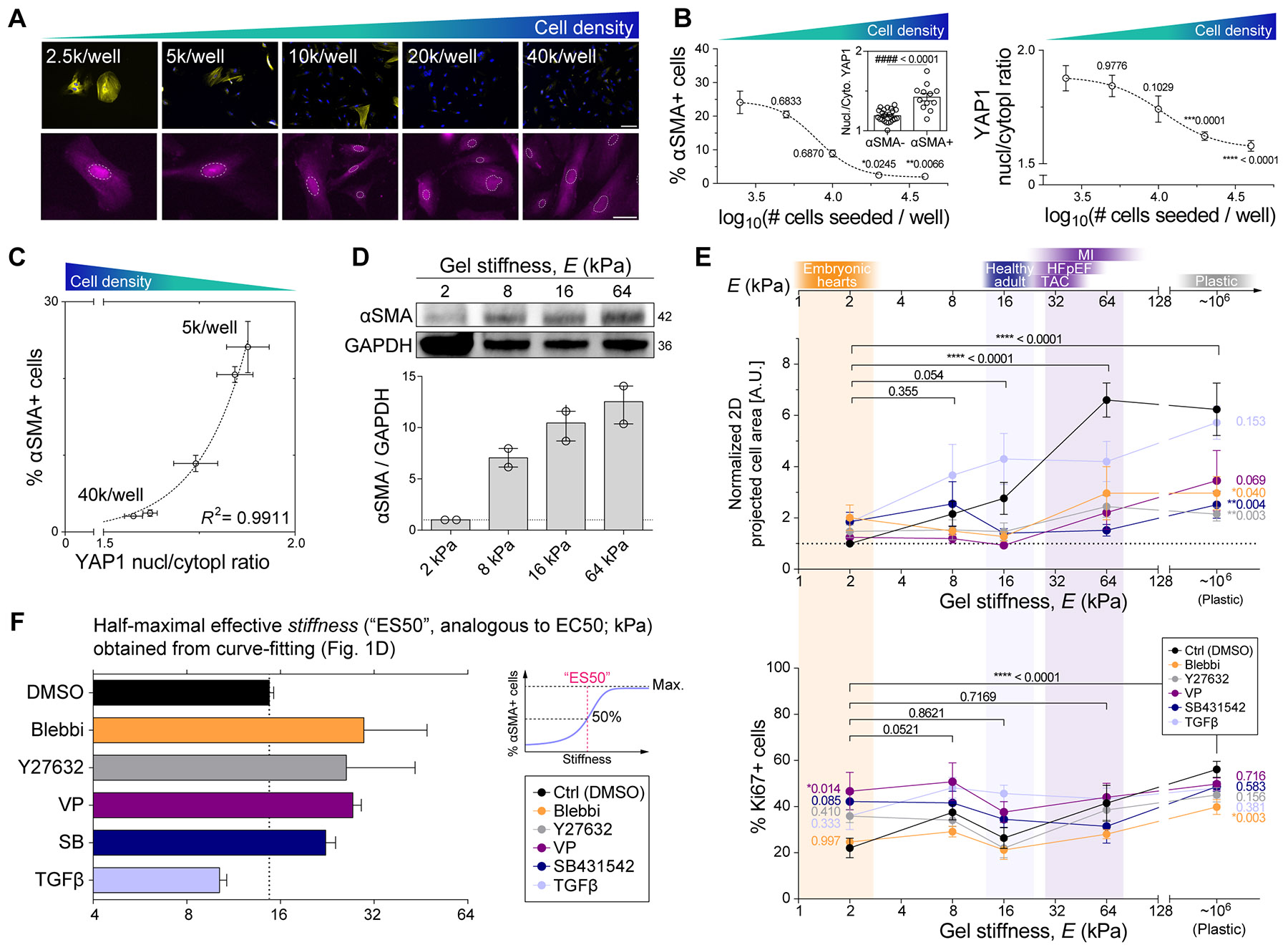
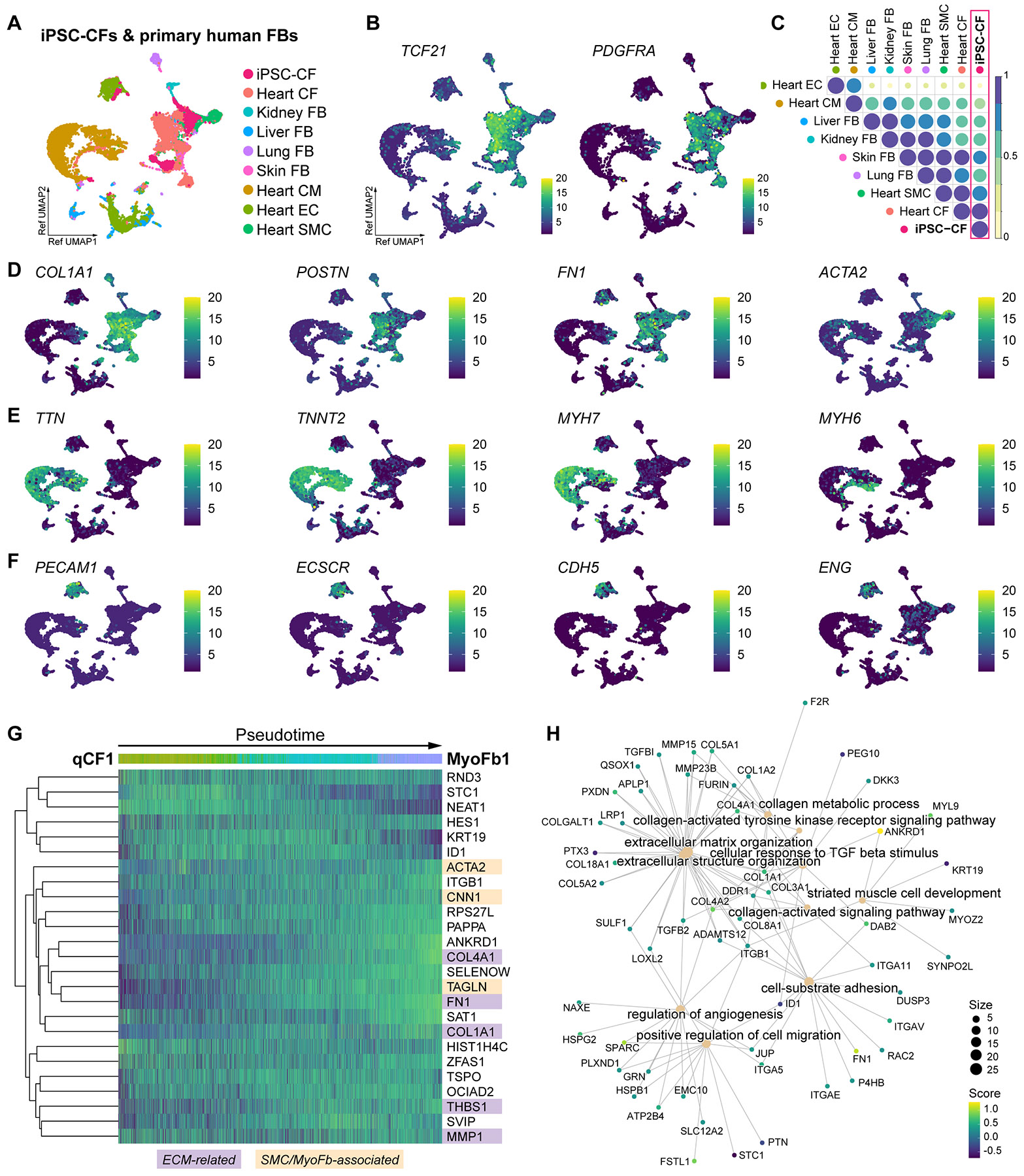
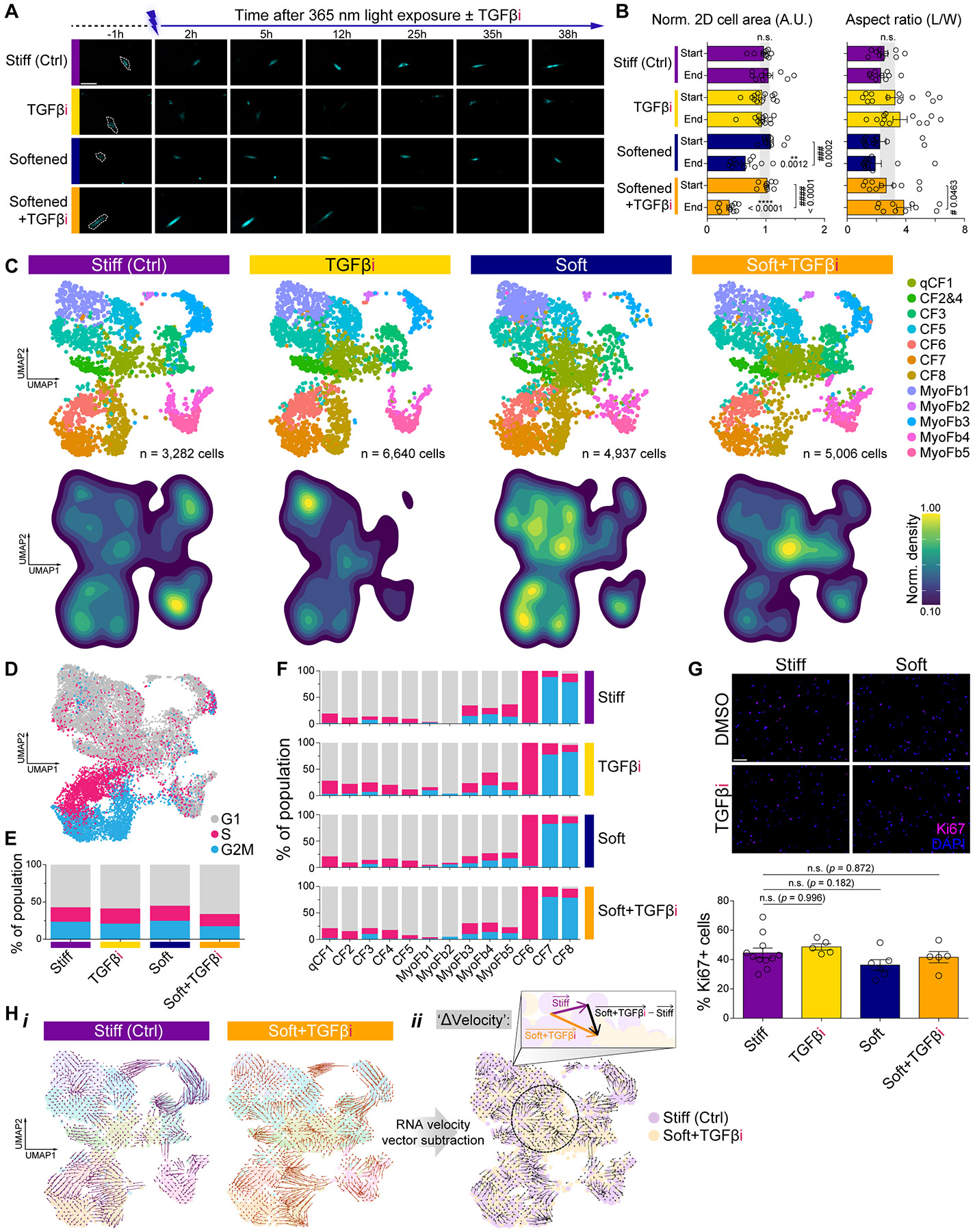
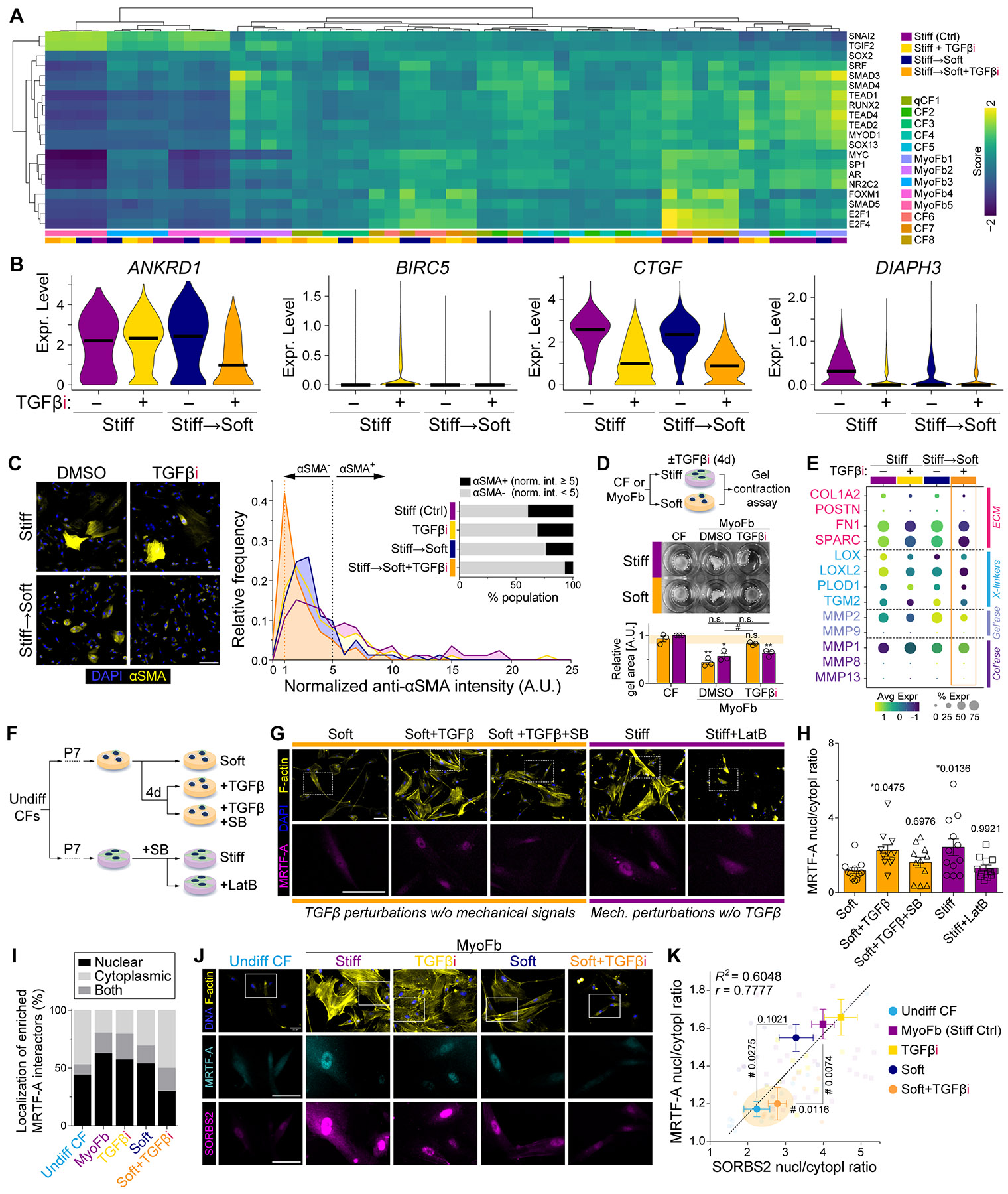
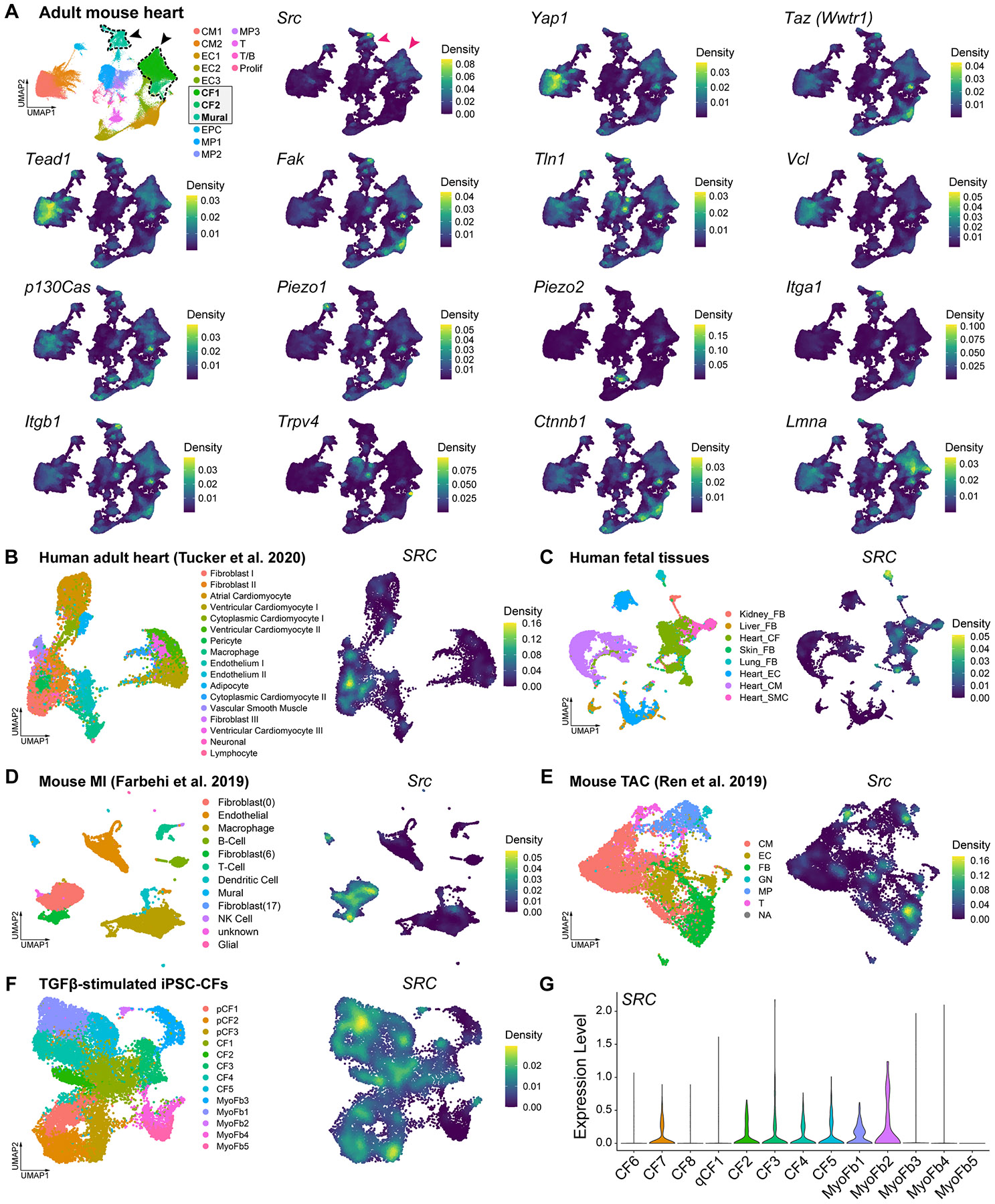
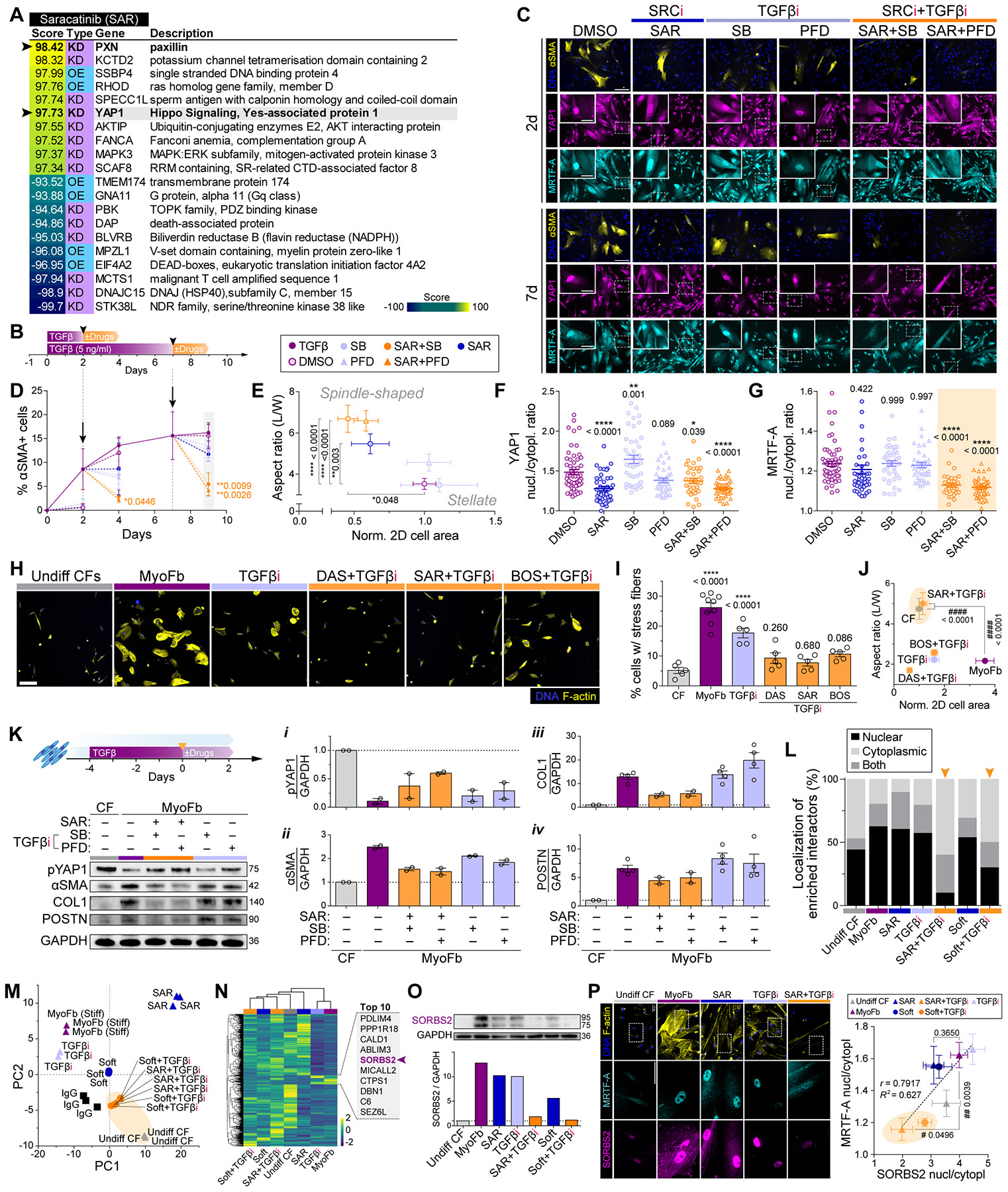
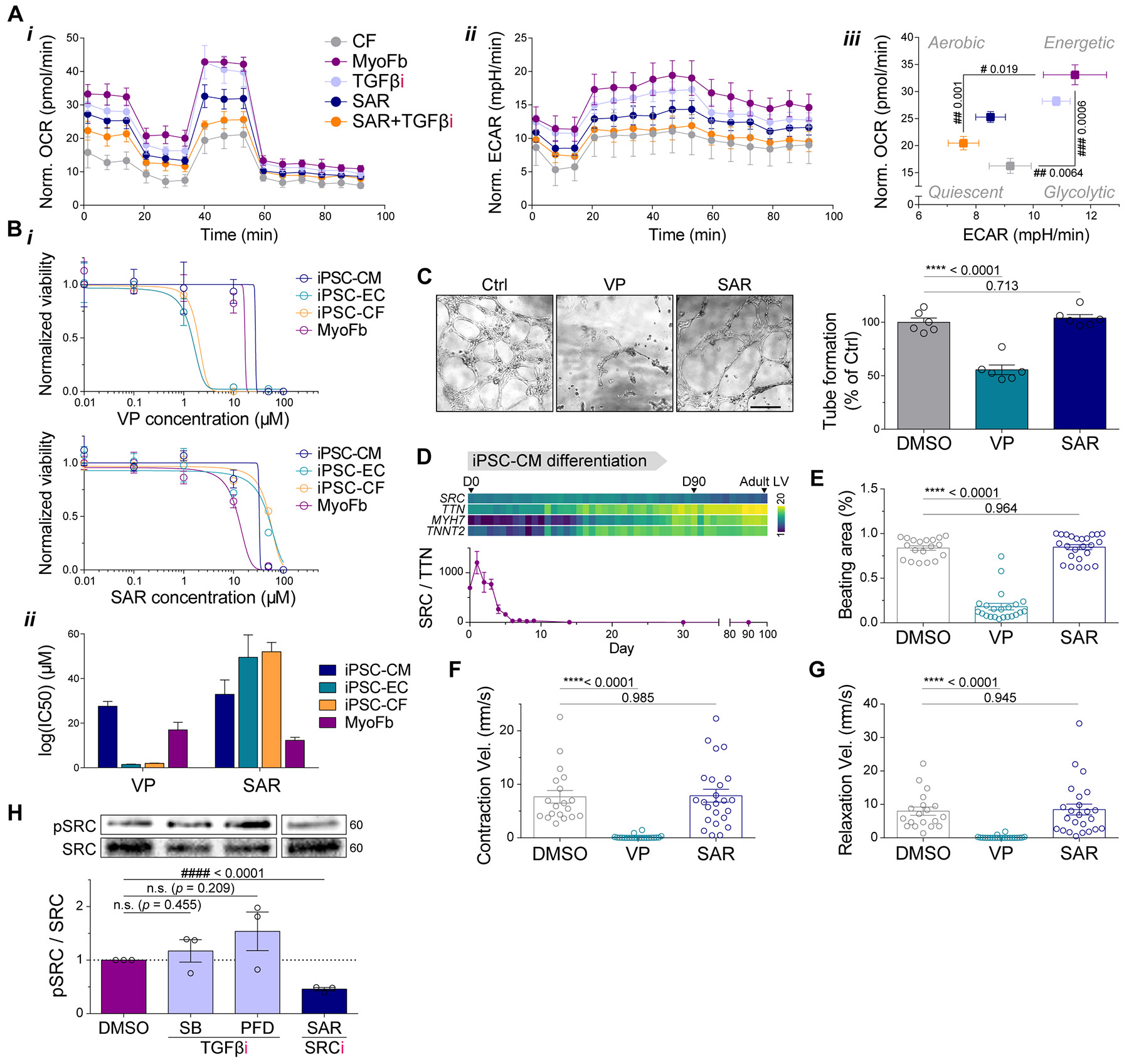
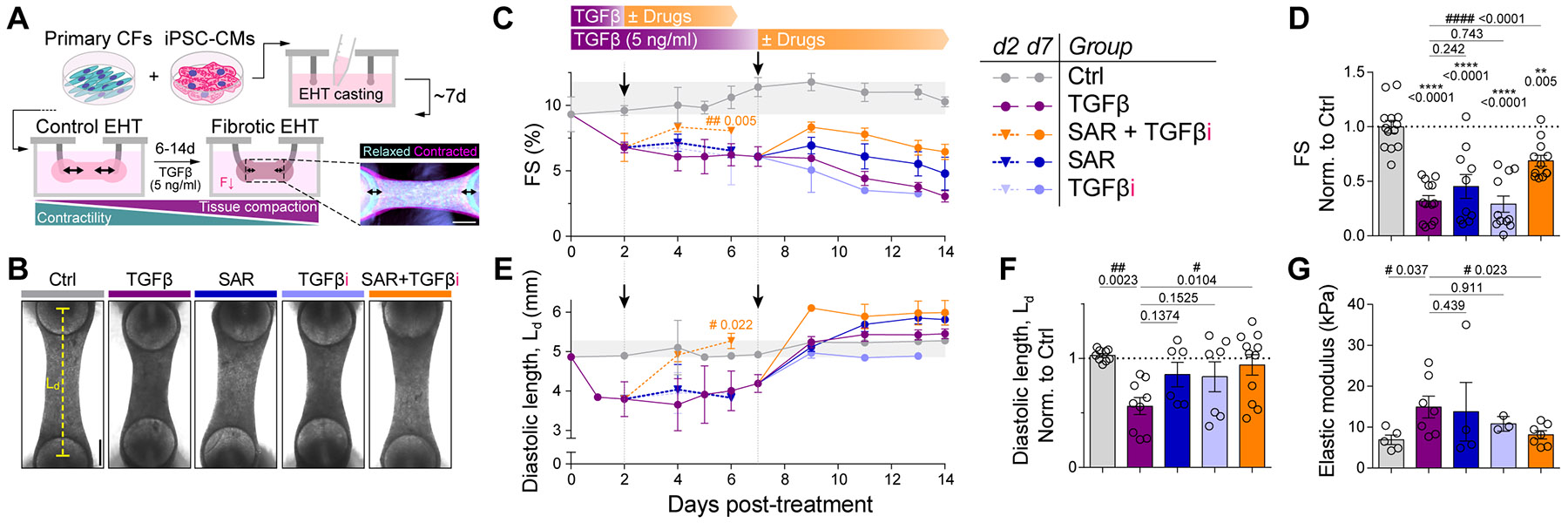
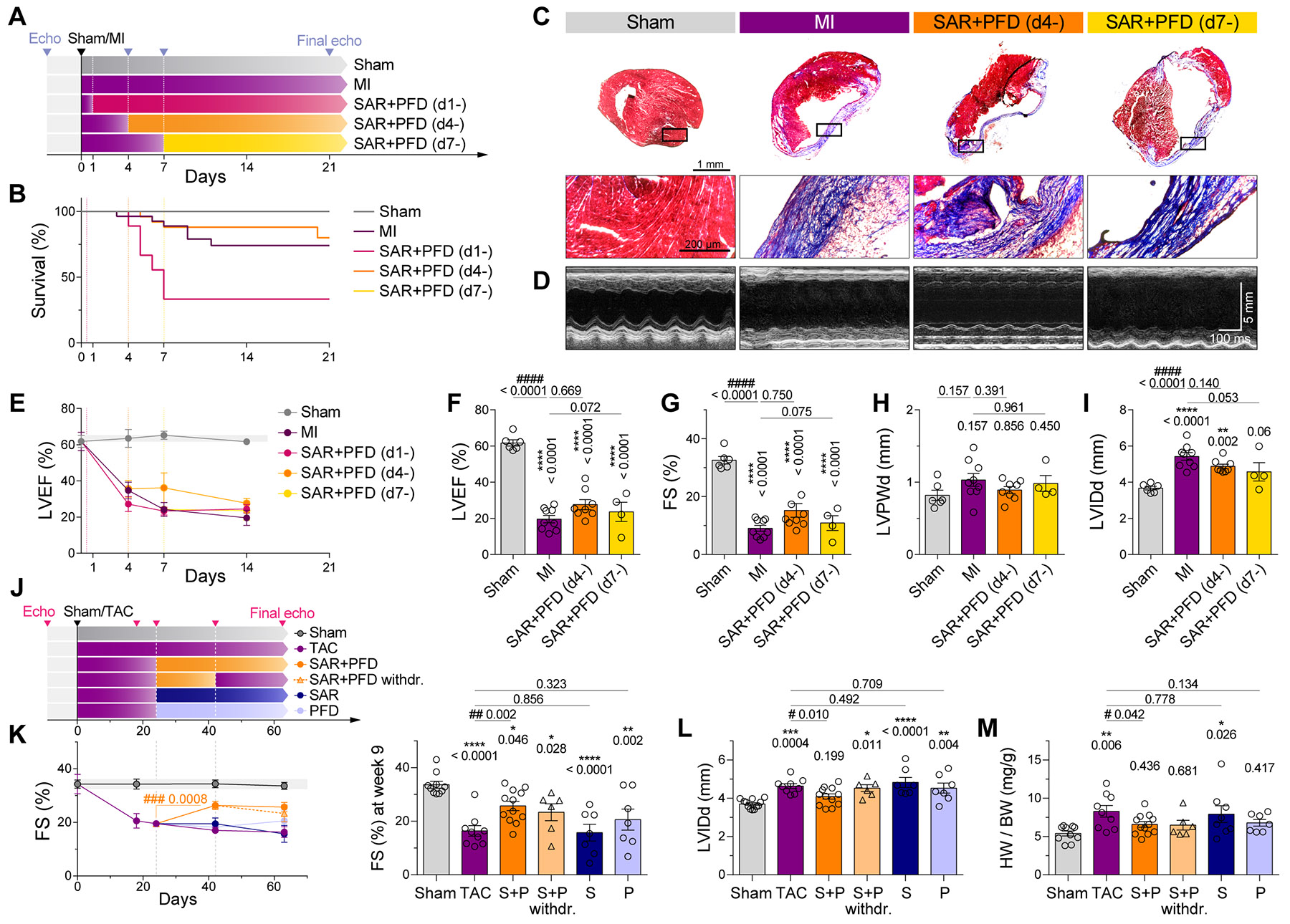
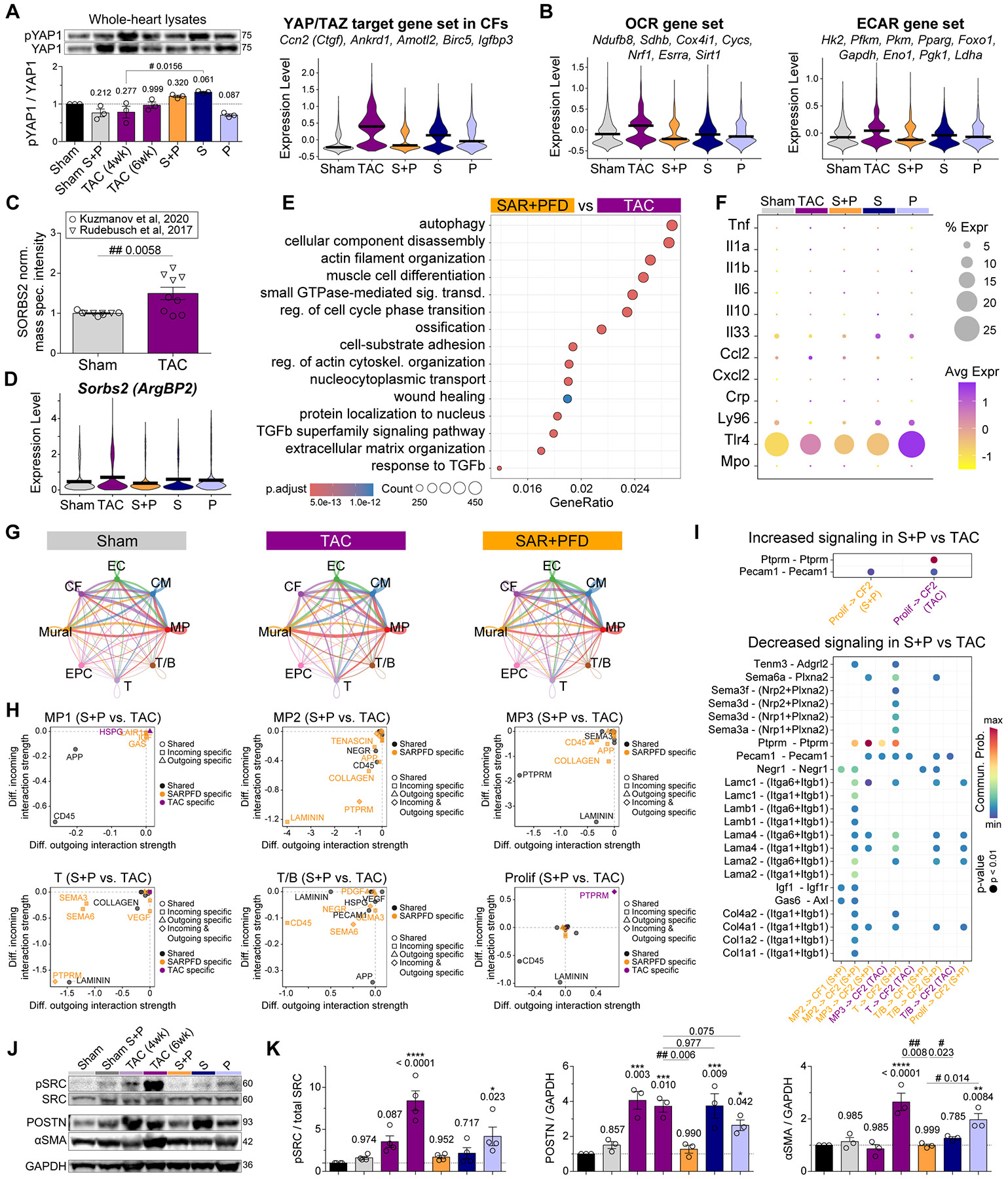
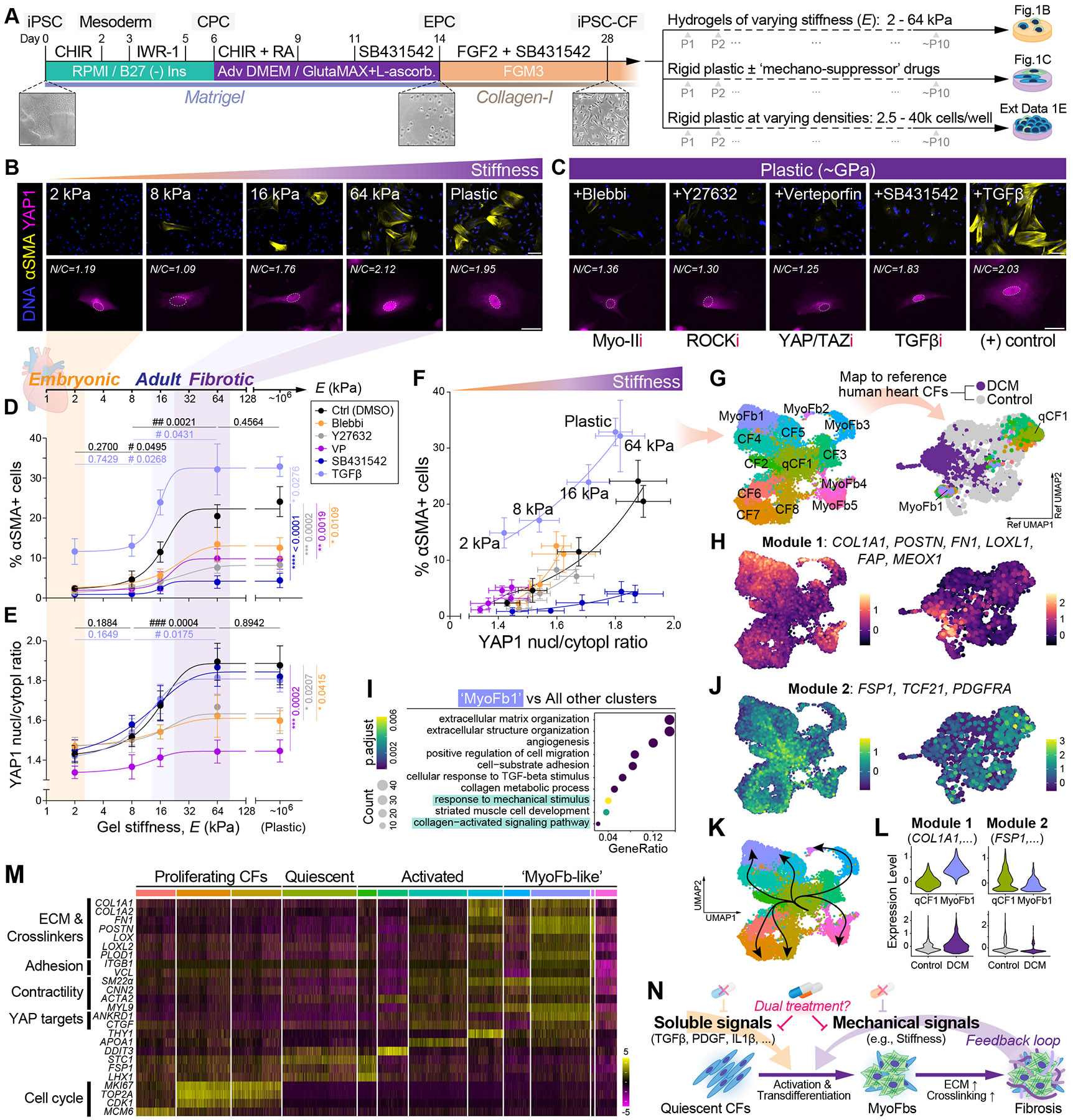
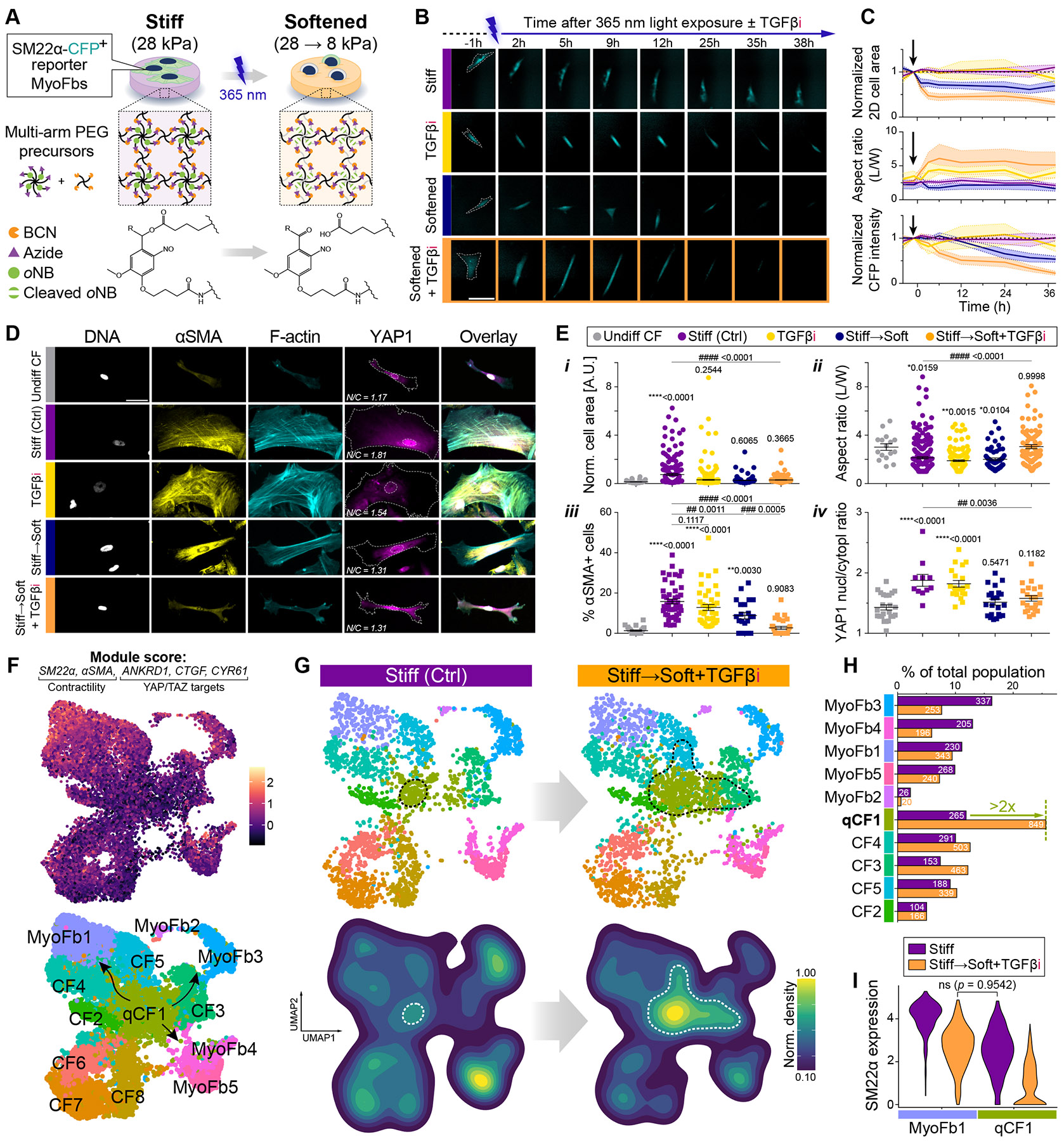
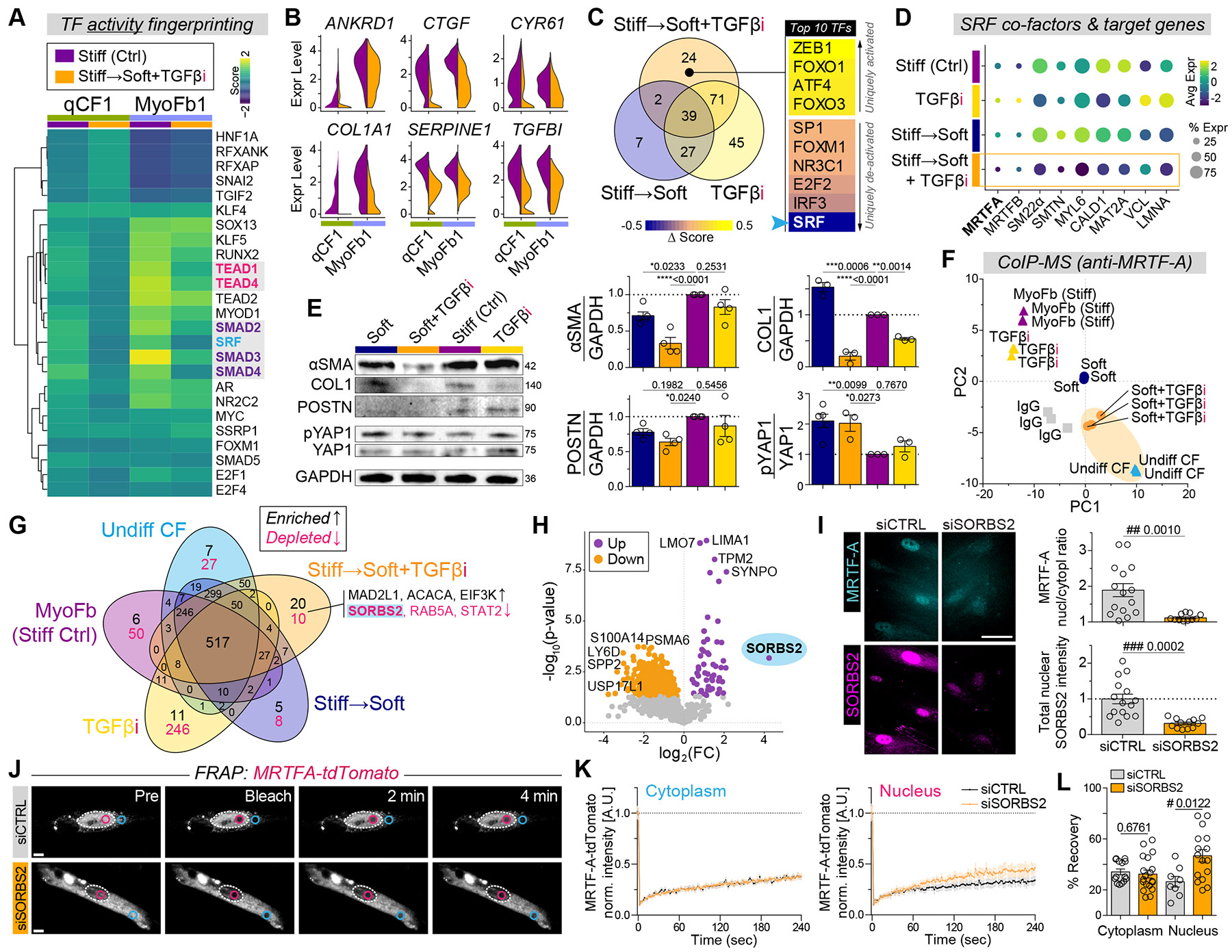
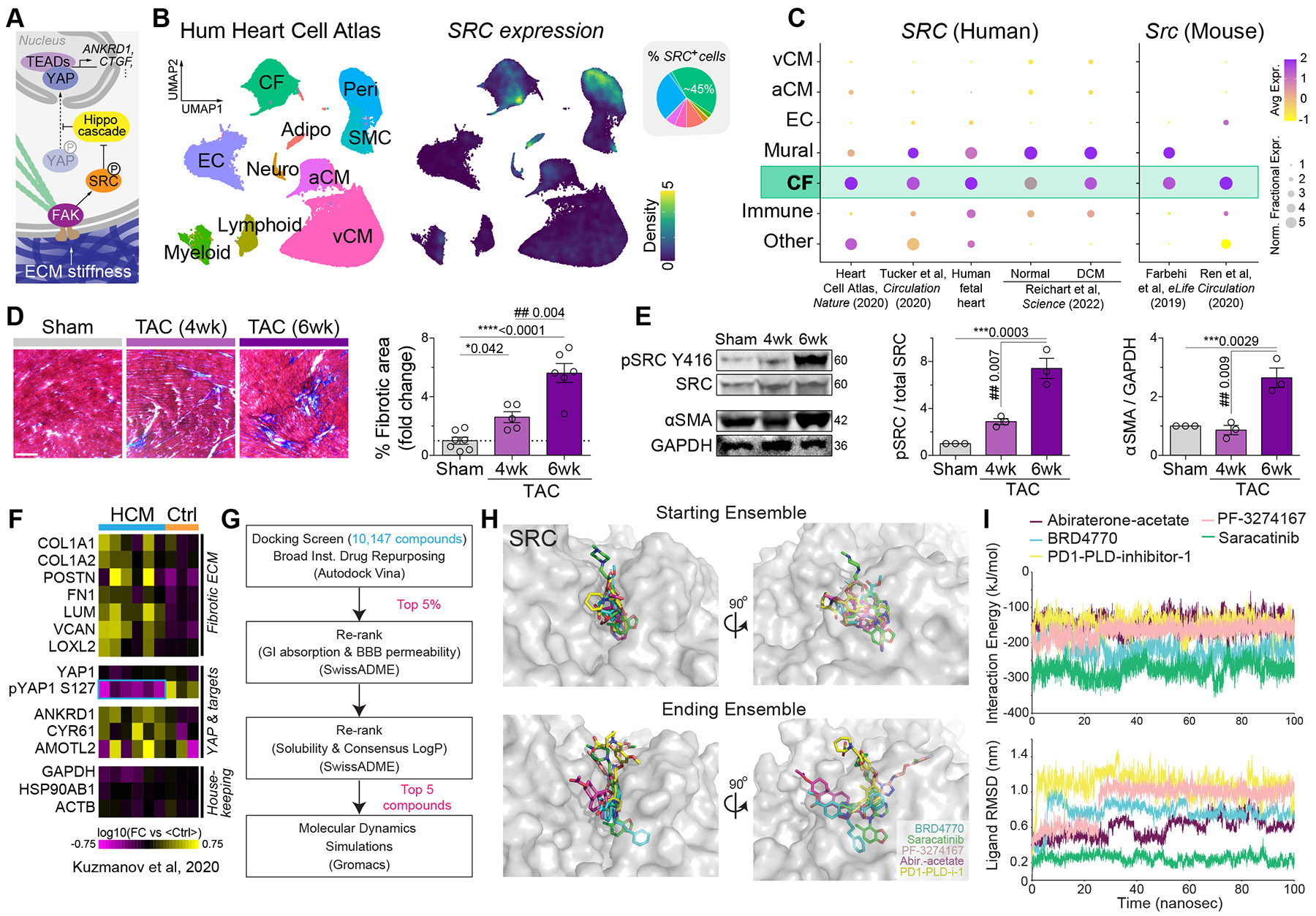
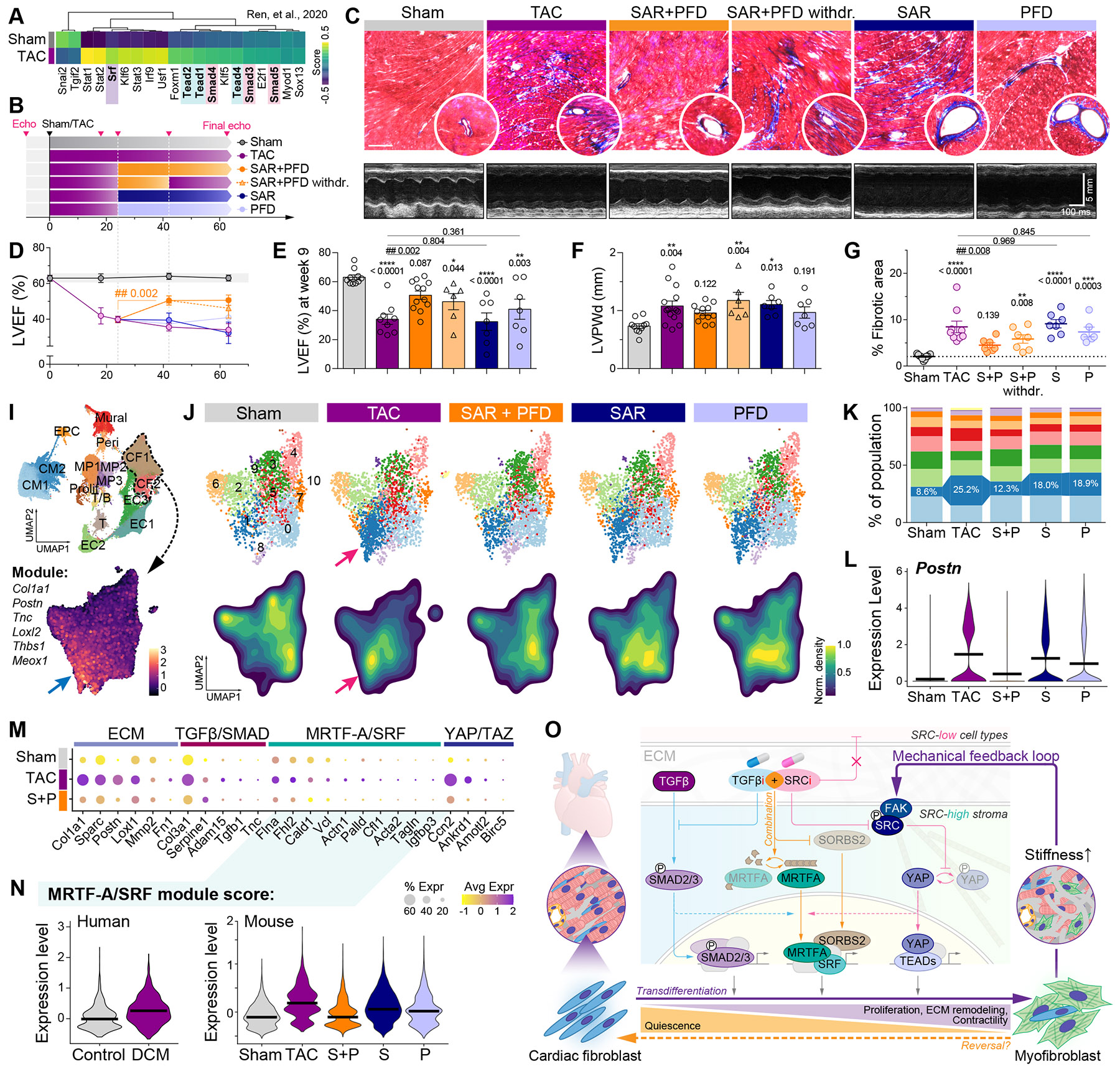
Matrix-derived biophysical cues are known to regulate the activation of fibroblasts and their subsequent transdifferentiation into myofibroblasts1-6, but whether modulation of these signals can suppress fibrosis in intact tissues remains unclear, particularly in the cardiovascular system7-10. Here we demonstrate across multiple scales that inhibition of matrix mechanosensing in persistently activated cardiac fibroblasts potentiates-in concert with soluble regulators of the TGFβ pathway-a robust transcriptomic, morphological and metabolic shift towards quiescence. By conducting a meta-analysis of public human and mouse single-cell sequencing datasets, we identify the focal-adhesion-associated tyrosine kinase SRC as a fibroblast-enriched mechanosensor that can be targeted selectively in stromal cells to mimic the effects of matrix softening in vivo. Pharmacological inhibition of SRC by saracatinib, coupled with TGFβ suppression, induces synergistic repression of key profibrotic gene programs in fibroblasts, characterized by a marked inhibition of the MRTF-SRF pathway, which is not seen after treatment with either drug alone. Importantly, the dual treatment alleviates contractile dysfunction in fibrotic engineered heart tissues and in a mouse model of heart failure. Our findings point to joint inhibition of SRC-mediated stromal mechanosensing and TGFβ signalling as a potential mechanotherapeutic strategy for treating cardiovascular fibrosis.
© 2025. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests: J.C.W. is a co-founder and member of the scientific advisory board of Greenstone Biosciences. H.M.B. is a cofounder and member of the scientific advisory board of Epirium Bio. The other authors declare no competing interests.
Figures
References
References (Materials and Methods)
MeSH terms
Substances
Grants and funding
- R01 HL163680/HL/NHLBI NIH HHS/United States
- F32 HL152483/HL/NHLBI NIH HHS/United States
- R01 HL130020/HL/NHLBI NIH HHS/United States
- R01 HL146690/HL/NHLBI NIH HHS/United States
- R01 HL141371/HL/NHLBI NIH HHS/United States
- R01 HL150693/HL/NHLBI NIH HHS/United States
- K99 HL166695/HL/NHLBI NIH HHS/United States
- K99 HL163443/HL/NHLBI NIH HHS/United States
- F32 HL173968/HL/NHLBI NIH HHS/United States
- R01 HL113006/HL/NHLBI NIH HHS/United States
- R01 HL145676/HL/NHLBI NIH HHS/United States
- R01 HL141851/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
