

Updated insights into the molecular networks for NLRP3 inflammasome activation
- PMID: 40307577
- PMCID: PMC12125403
- DOI: 10.1038/s41423-025-01284-9
Updated insights into the molecular networks for NLRP3 inflammasome activation
Abstract
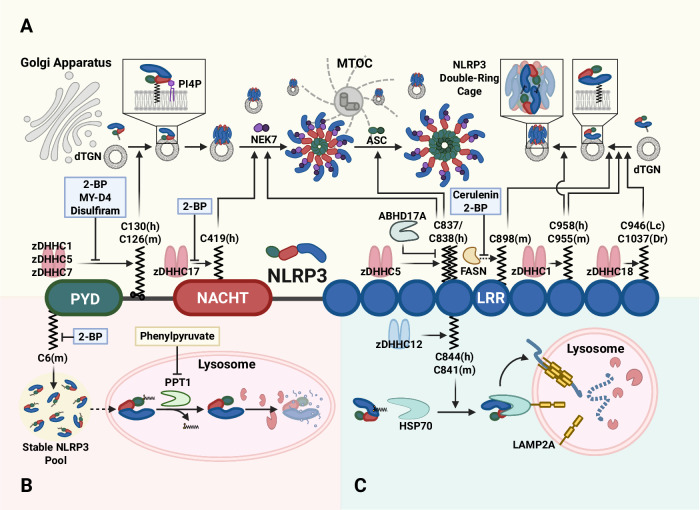
Over the past decade, significant advances have been made in our understanding of how NACHT-, leucine-rich-repeat-, and pyrin domain-containing protein 3 (NLRP3) inflammasomes are activated. These findings provide detailed insights into the transcriptional and posttranslational regulatory processes, the structural-functional relationship of the activation processes, and the spatiotemporal dynamics of NLRP3 activation. Notably, the multifaceted mechanisms underlying the licensing of NLRP3 inflammasome activation constitute a focal point of intense research. Extensive research has revealed the interactions of NLRP3 and its inflammasome components with partner molecules in terms of positive and negative regulation. In this Review, we provide the current understanding of the complex molecular networks that play pivotal roles in regulating NLRP3 inflammasome priming, licensing and assembly. In addition, we highlight the intricate and interconnected mechanisms involved in the activation of the NLRP3 inflammasome and the associated regulatory pathways. Furthermore, we discuss recent advances in the development of therapeutic strategies targeting the NLRP3 inflammasome to identify potential therapeutics for NLRP3-associated inflammatory diseases. As research continues to uncover the intricacies of the molecular networks governing NLRP3 activation, novel approaches for therapeutic interventions against NLRP3-related pathologies are emerging.
Keywords: Inflammatory disease; Licensing; NLRP3 inflammasome; Post-translational modification (PTM); Pyroptosis; Spatiotemporal.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
