

Endoplasmic reticulum stress disrupts signaling via altered processing of transmembrane receptors
- PMID: 40307870
- PMCID: PMC12044870
- DOI: 10.1186/s12964-025-02208-w
Endoplasmic reticulum stress disrupts signaling via altered processing of transmembrane receptors
Abstract
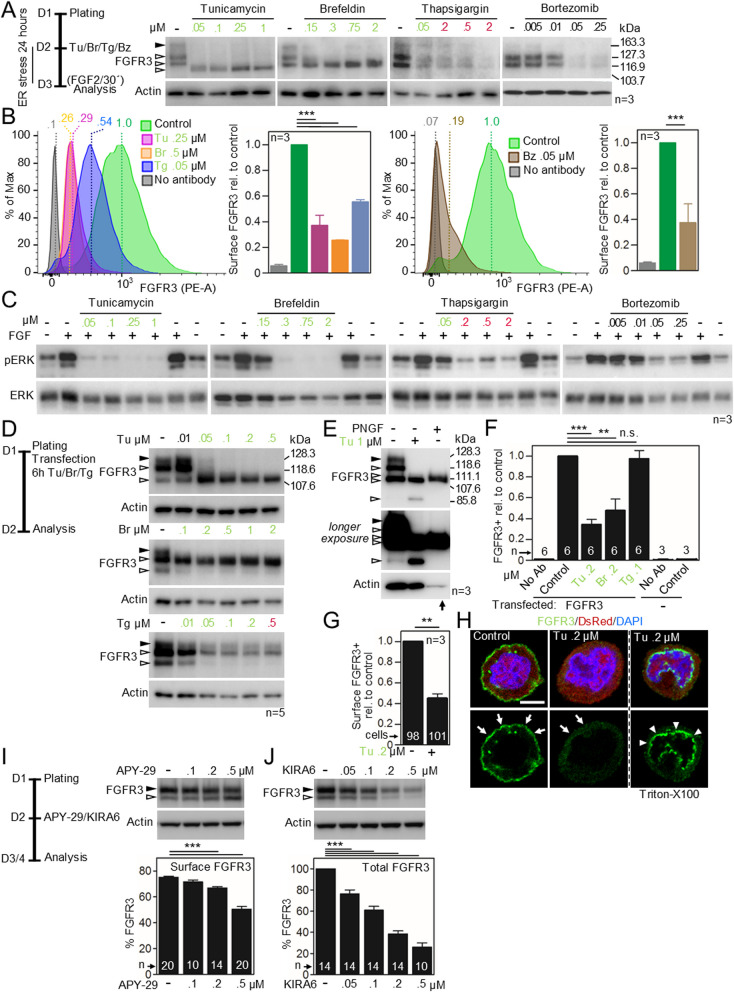
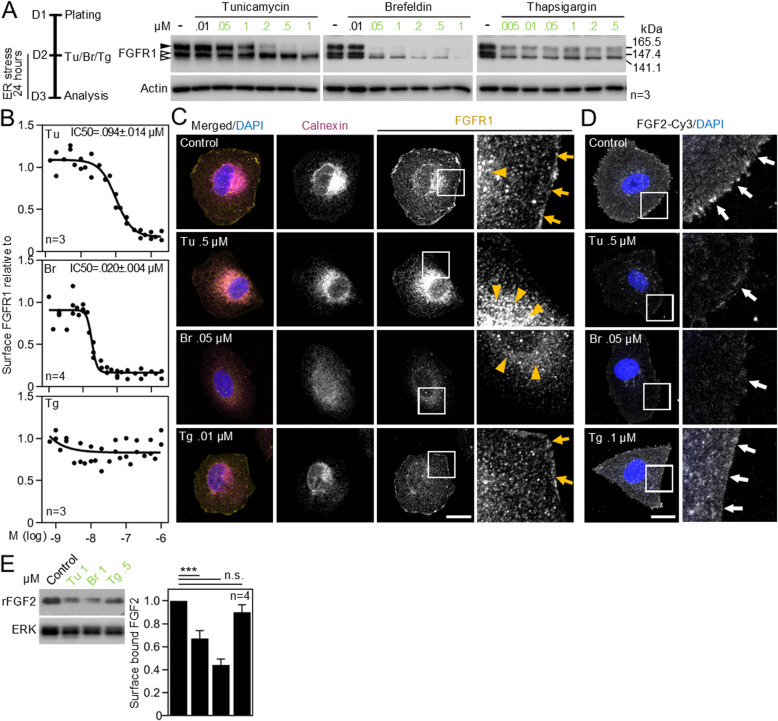
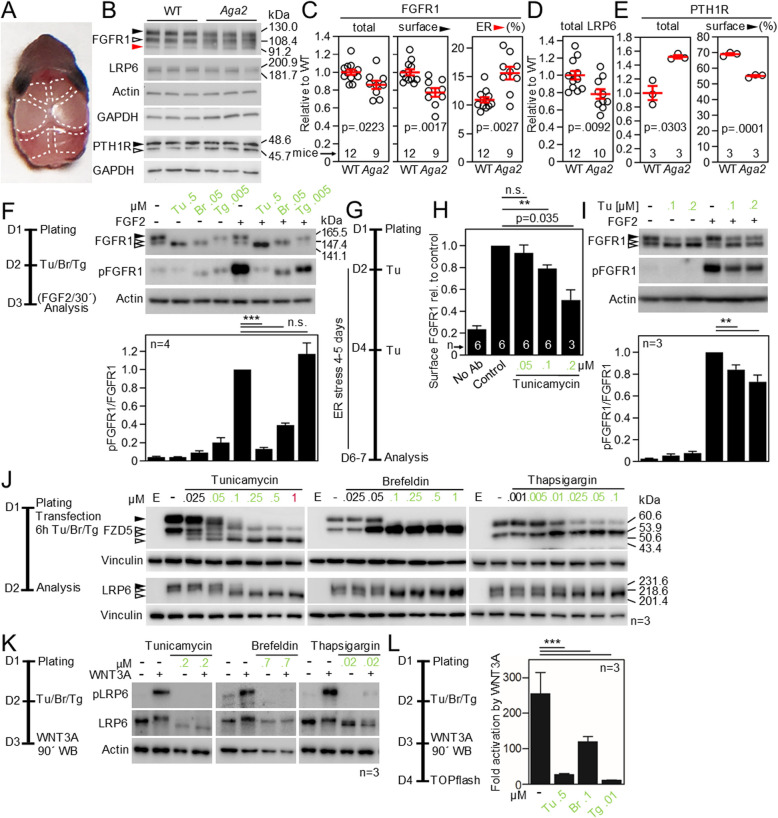
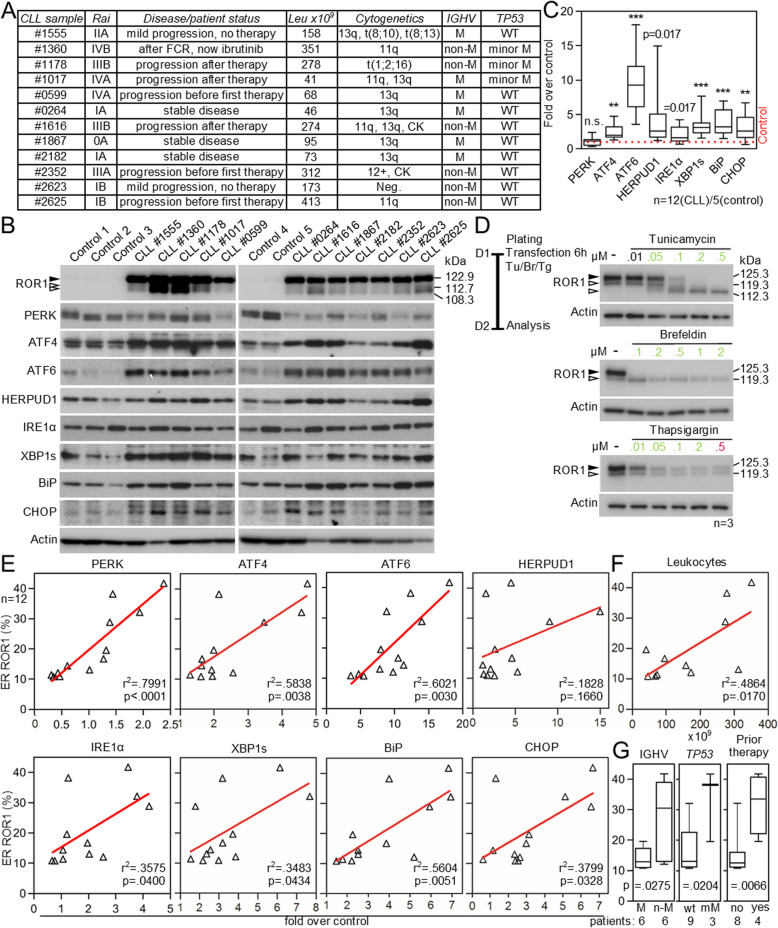
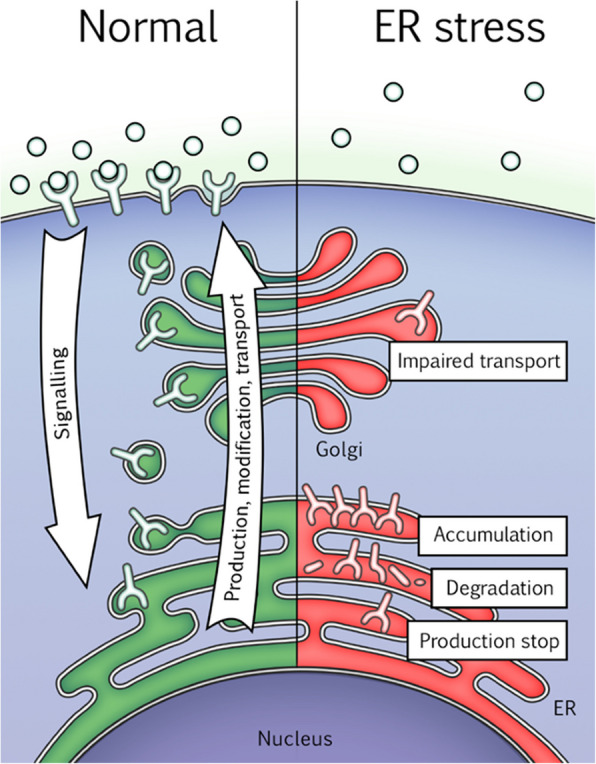
Cell communication systems based on polypeptide ligands use transmembrane receptors to transmit signals across the plasma membrane. In their biogenesis, receptors depend on the endoplasmic reticulum (ER)-Golgi system for folding, maturation, transport and localization to the cell surface. ER stress, caused by protein overproduction and misfolding, is a well-known pathology in neurodegeneration, cancer and numerous other diseases. How ER stress affects cell communication via transmembrane receptors is largely unknown. In disease models of multiple myeloma, chronic lymphocytic leukemia and osteogenesis imperfecta, we show that ER stress leads to loss of the mature transmembrane receptors FGFR3, ROR1, FGFR1, LRP6, FZD5 and PTH1R at the cell surface, resulting in impaired downstream signaling. This is caused by downregulation of receptor production and increased intracellular retention of immature receptor forms. Reduction of ER stress by treatment of cells with the chemical chaperone tauroursodeoxycholic acid or by expression of the chaperone protein BiP resulted in restoration of receptor maturation and signaling. We show a previously unappreciated pathological effect of ER stress; impaired cellular communication due to altered receptor processing. Our findings have implications for disease mechanisms related to ER stress and are particularly important when receptor-based pharmacological approaches are used for treatment.
Keywords: ER; Endoplasmic reticulum; Impaired; Receptor; Signaling; Stress; Transmembrane.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures

References
-
- Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–97. - PubMed
-
- Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–206. - PubMed
-
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. - PubMed
MeSH terms
Substances
Grants and funding
- NIH AR 006124, NIH P01 HD070394/GF/NIH HHS/United States
- CZ.02.2.69/0.0/0.0/19_073/0016943/IGA MUNI
- FNBr, 65269705/MH CZ - DRO
- Geisman award/Osteogenesis Imperfecta Foundation
- GA21-11585S/Czech Science Foundation
- Praemium Academiae/Czech Academy of Sciences
- LUAUS23295/Ministry of Education, Youth and Sports of the Czech Republic
- GF21-26400K/Czech Science Foundation
- NU23-10-00550/Agency for Healthcare Research of the Czech Republic
- P01 HD070394/HD/NICHD NIH HHS/United States
- Programme EXCELES, ID Project No. LX22NPO5102/RC/CCR NIH HHS/United States
- T32 GM152342/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
