

Head and neck squamous cell carcinoma-derived extracellular vesicles mediate Ca²⁺-dependent platelet activation and aggregation through tissue factor
- PMID: 40312345
- PMCID: PMC12044835
- DOI: 10.1186/s12964-025-02215-x
Head and neck squamous cell carcinoma-derived extracellular vesicles mediate Ca²⁺-dependent platelet activation and aggregation through tissue factor
Abstract
Background: Head and neck squamous cell carcinoma (HNSCC) is an aggressive malignancy, characterized by poor clinical outcomes, primarily driven by high rate of locoregional recurrence and metastasis. Extensive heterogeneity among the tumor cells as well as modulation of a highly immunosuppressive tumor microenvironment shape cancer progression. Shedding of extracellular vesicles (EVs) derived from tumor cells is a critical mediator of the disease initiating horizontal transfer of tumor components into platelets. This triggers platelet activation and thromboinflammation fueling tumor progression through multiple mechanisms.
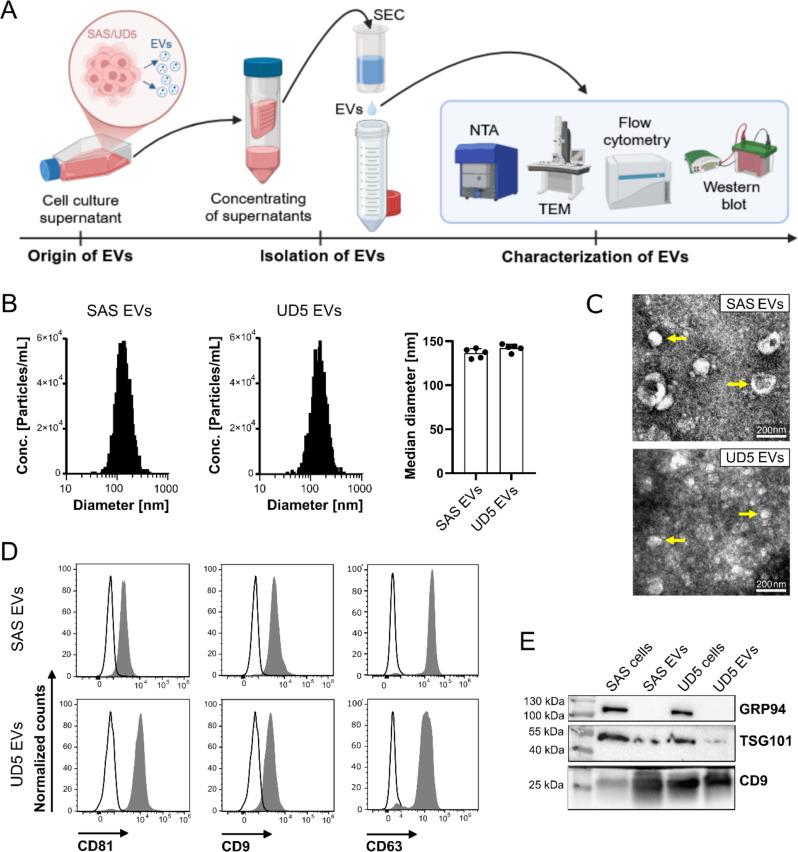
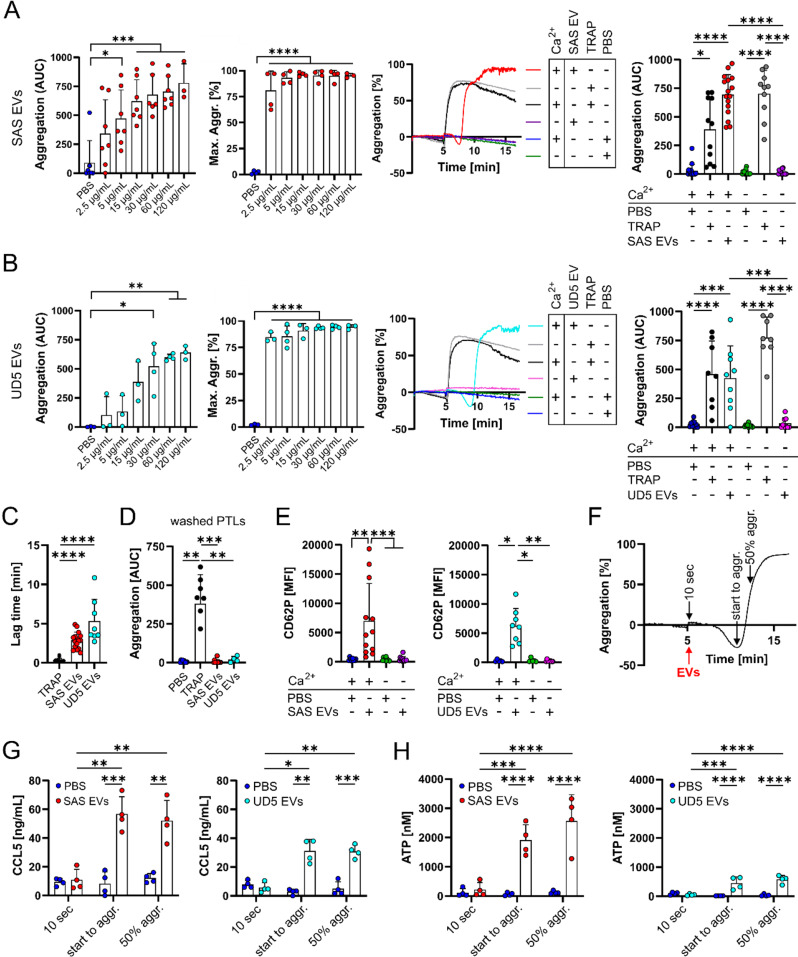
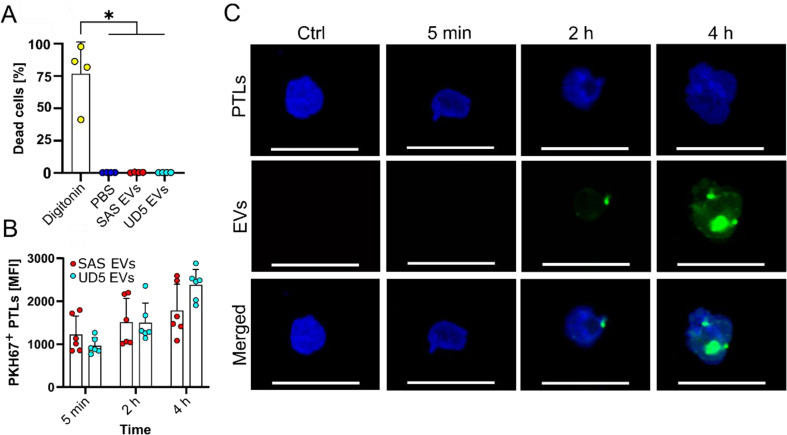
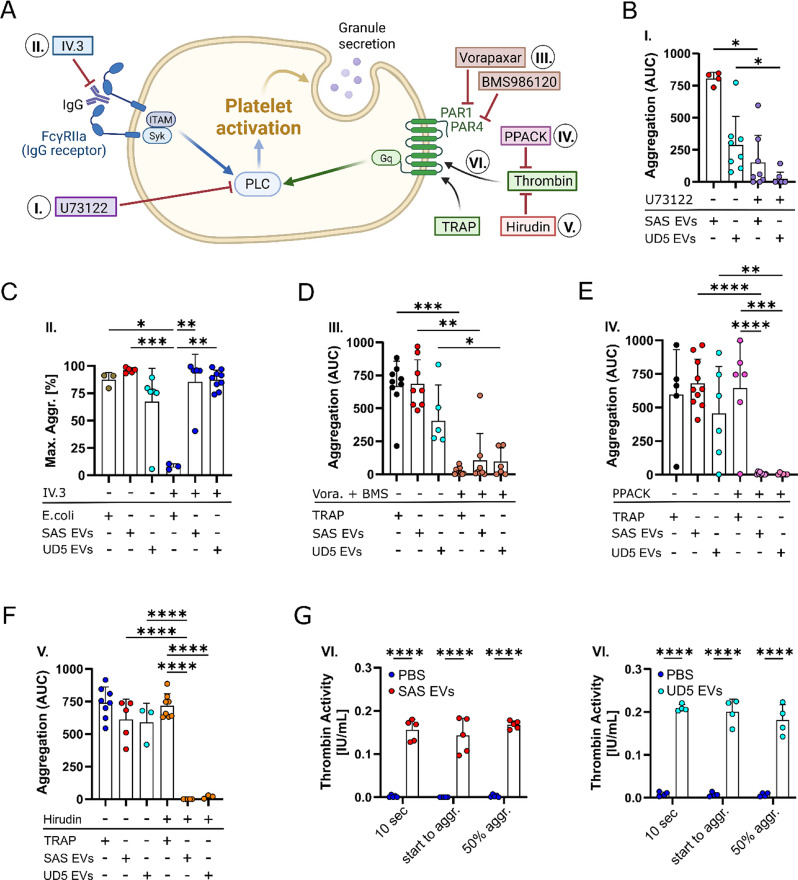
Methods: HNSCC-derived EVs isolated from HNSCC cell lines (SAS, UD-SCC 5) using size exclusion chromatography and characterized via flow cytometry, electron microscopy, nanoparticle tracking analysis and Western blotting, were used to induce platelet activation and aggregation, measured by aggregometry, flow cytometry, as well as the release of chemokines and Adenosine triphosphate, which were quantified using enzyme-linked immunosorbent assays (ELISA). Mechanistic investigations included inhibitor assays, thrombin activity measurements, and proteomic analyses.
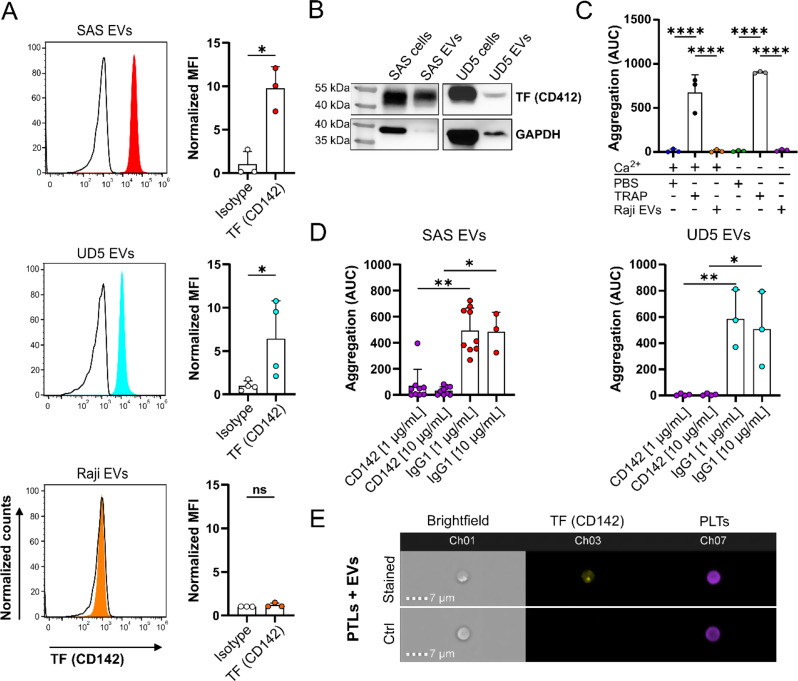
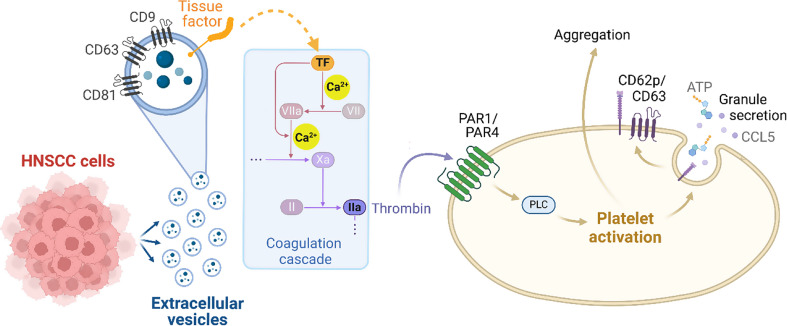
Results: We could show that EVs do not activate platelets through the FcγRIIa-IgG axis but platelet activation and aggregation is induced in a calcium-dependent manner, primarily mediated by EV-associated tissue factor. Proteomic analysis confirmed the presence of tissue factor in these vesicles, implicating its involvement in initiating the coagulation cascade, that leads to platelet activation and aggregation. This process was characterized by delayed aggregation kinetics and relied on thrombin activation, as the inhibition of thrombin and its receptors reduced platelet aggregation. HNSCC-derived EVs are pivotal in establishing a prothrombotic environment by promoting platelet activation and aggregation through tissue factor-dependent thrombin generation.
Conclusion: These findings indicate a therapeutic potential of targeting EV-mediated pathways as a therapeutic approach to alleviate thrombotic complications in HNSCC patients. Subsequent animal studies will be crucial to validate and extend these observations, providing deeper insight into their clinical implications.
Keywords: Cancer-associated thrombosis; Coagulation cascade; Extracellular vesicles; Head and neck squamous cell carcinoma; Platelet activation; Tissue factor.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study was approved by the local ethics committee of the Technical University of Munich (protocol numbers: 2020 − 474_2-S-NP and 2022-211-S-NP). All donors provided informed consent in compliance with the Declaration of Helsinki. Donors were self-reported as healthy and confirmed they had not taken antithrombotic medication for at least 7 days prior to blood donation. Competing interests: The authors declare no competing interests.
Figures
References
-
- Lala M, Chirovsky D, Cheng JD, Mayawala K. Clinical outcomes with therapies for previously treated recurrent/metastatic head-and-neck squamous cell carcinoma (R/M HNSCC): A systematic literature review. Oral Oncol. 2018;84:108–20. - PubMed
-
- Wang G, Zhang M, Cheng M, Wang X, Li K, Chen J, et al. Tumor microenvironment in head and neck squamous cell carcinoma: functions and regulatory mechanisms. Cancer Lett. 2021;507:55–69. - PubMed
-
- Xu Q, Li X. Tumor-derived extracellular vesicles in the immune microenvironment of head and neck squamous cell carcinoma: foe or future? J Stomatology Oral Maxillofacial Surg. 2024;125(4):101738. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
