

Estrogen-secreting testicular tumors in 46,XY female patients with 17α-hydroxylase/17,20-lyase deficiency: two unusual case reports and a review of the literature
- PMID: 40313596
- PMCID: PMC12043694
- DOI: 10.3389/fgene.2025.1508792
Estrogen-secreting testicular tumors in 46,XY female patients with 17α-hydroxylase/17,20-lyase deficiency: two unusual case reports and a review of the literature
Abstract
Context: 17α-hydroxylase/17,20-lyase deficiency (17OHD) is a rare autosomal recessive condition. Women who have the complete form of 17OHD typically have a female phenotype, with an absence of secondary sexual characteristics, primary amenorrhea, and hypertension, which is usually detected in adolescence. Generally, 46,XY patients with a partial form of 17OHD have atypical genitalia and intra-abdominal or inguinal testes. The risk of developing malignant testicular tumors or testicular adrenal rest tumors in 21-hydroxylase deficiency congenital adrenal hyperplasia is reported in 46,XY patients. In contrast, these conditions are rarely described in patients with 17OHD.
Objective: This study aims to investigate patients with 17OHD who exhibit testicular tumors and spontaneous pubertal development.
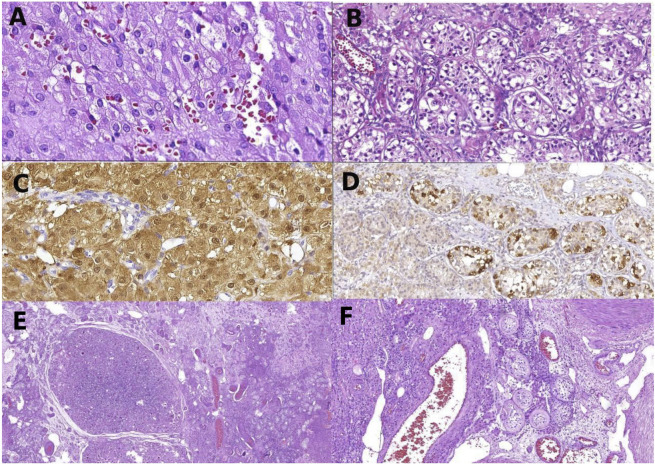
Patients and results: Two unrelated women with 46,XY karyotype with 17OHD who presented with unexpected spontaneous development of secondary sexual characteristics and testicular tumors were described. Pathogenic allelic variants in CYP17A1 were identified in the compound heterozygous state in both patients. The variants p.Trp406Arg and p.Pro428Leu were identified in the patient with Leydig cell neoplasia plus germ cell neoplasia in situ, and the p.Arg358Gln and p.Trp406Arg variants were identified in the patient with intratubular seminoma associated with invasive classic seminoma.
Conclusion: Our findings reinforce the risk of testicular tumor development among 46,XY patients with 17OHD and add data to the discussion of the risk/benefit ratio of prophylactic gonadectomy in the treatment patients with 46, XY differences in sex development (DSD).
Keywords: 17α-hydroxylase/17; 20-lyase deficiency; 46,XY differences in sex development; CYP17A1 variants; Leydig cell tumor; seminoma; testicular tumor.
Copyright © 2025 Batatinha, Nishi, Batista, Faria Júnior, Sircili, Denes, Mesia, Salvanini, Costa, Carvalho, Mendonca and Domenice.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Barros B. A., Oliveira L. R. D., Surur C. R. C., Barros-Filho A. D. A., Maciel-Guerra A. T., Guerra-Junior G. (2021). Complete androgen insensitivity syndrome and risk of gonadal malignancy: systematic review. Ann Pediatr Endocrinol Metab. 31 março 26 (1), 19–23. 10.6065/apem.2040170.085 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources