

This is a preprint.
Harnessing the Power of Single-Cell Large Language Models with Parameter Efficient Fine-Tuning using scPEFT
- PMID: 40313770
- PMCID: PMC12045372
- DOI: 10.21203/rs.3.rs-5926885/v1
Harnessing the Power of Single-Cell Large Language Models with Parameter Efficient Fine-Tuning using scPEFT
Abstract
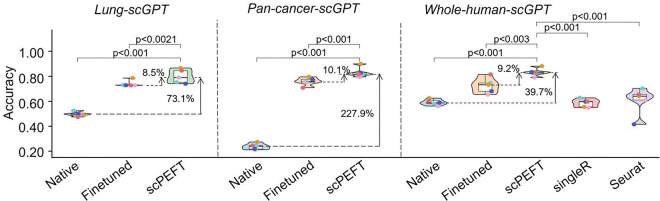
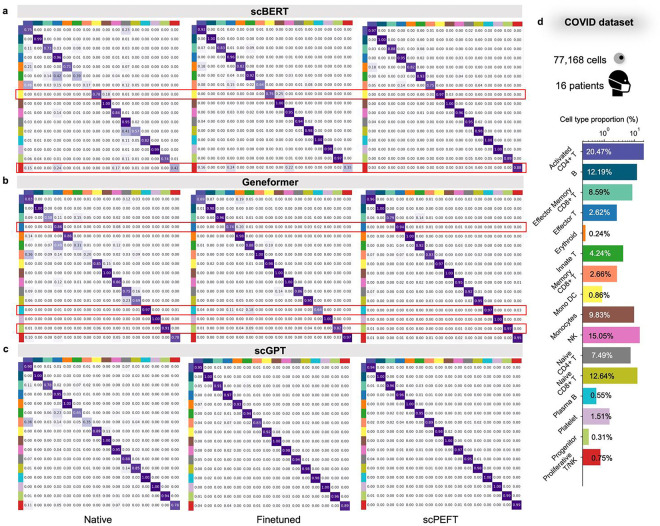
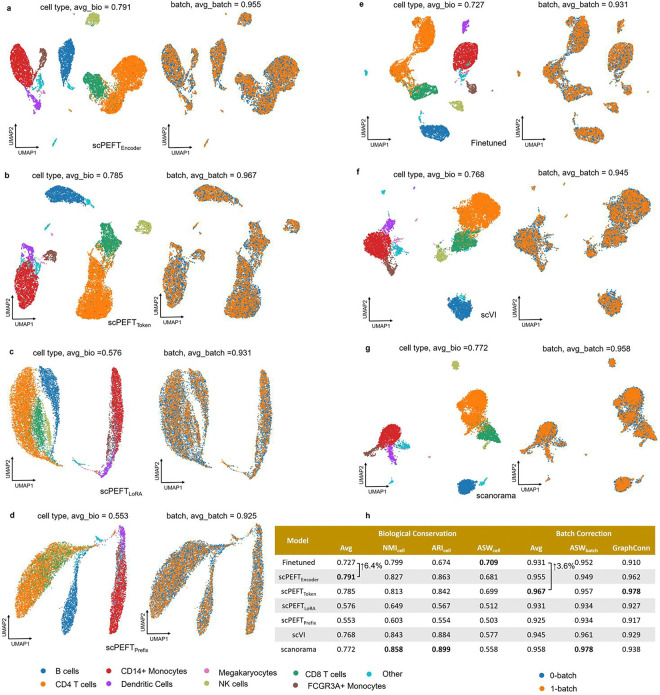
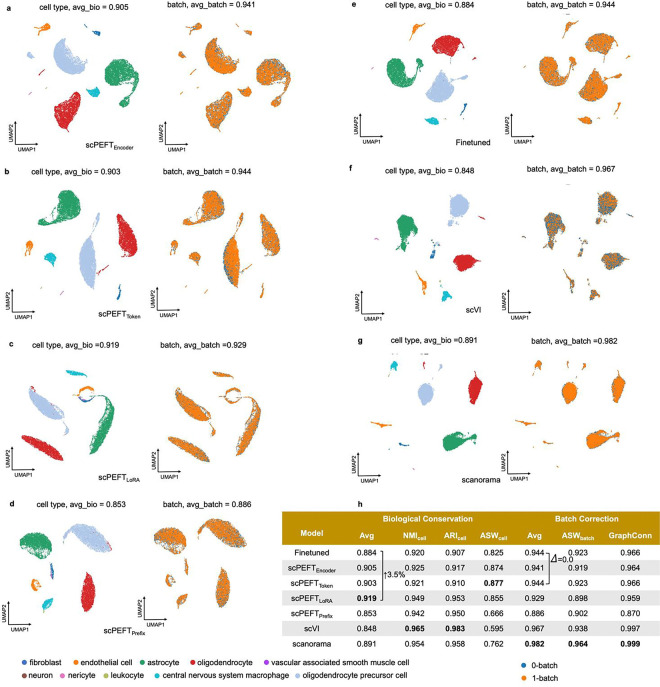
Single-cell large language models (scLLMs) capture essential biological insights from vast single-cell atlases but struggle in out-of-context applications, where zero-shot predictions can be unreliable. To address this, we introduce a single-cell parameter-efficient fine-tuning (scPEFT) framework that integrates learnable, low-dimensional adapters into scLLMs. By freezing the backbone model and updating only the adapter parameters, scPEFT efficiently adapts to specific tasks using limited custom data. This approach mitigates catastrophic forgetting, reduces parameter tuning by over 96%, and decreases GPU memory usage by more than half, significantly enhancing scLLMs's accessibility for resource-constrained researchers. Validated across diverse datasets, scPEFT outperformed zero-shot models and traditional fine-tuning in disease-specific, cross-species, and under-characterized cell population tasks. Its attention-mechanism analysis identified COVID-related genes associated with specific cell states and uncovered unique blood cell subpopulations, demonstrating scPEFT's capacity for condition-specific interpretations. These findings position scPEFT as an efficient solution for improving scLLMs' utilities in general single-cell analyses.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures




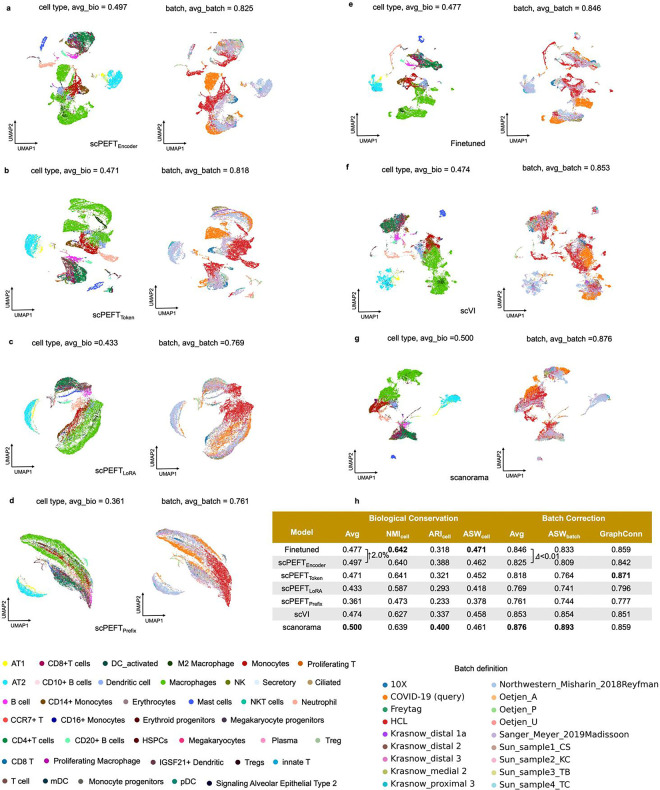
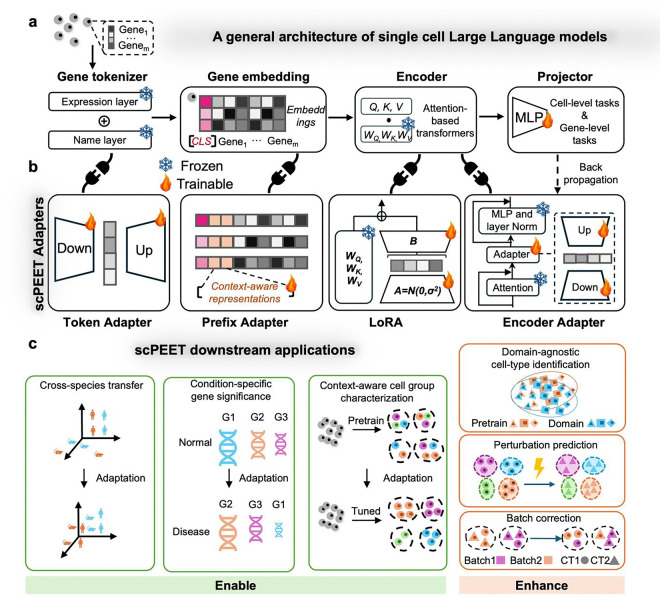
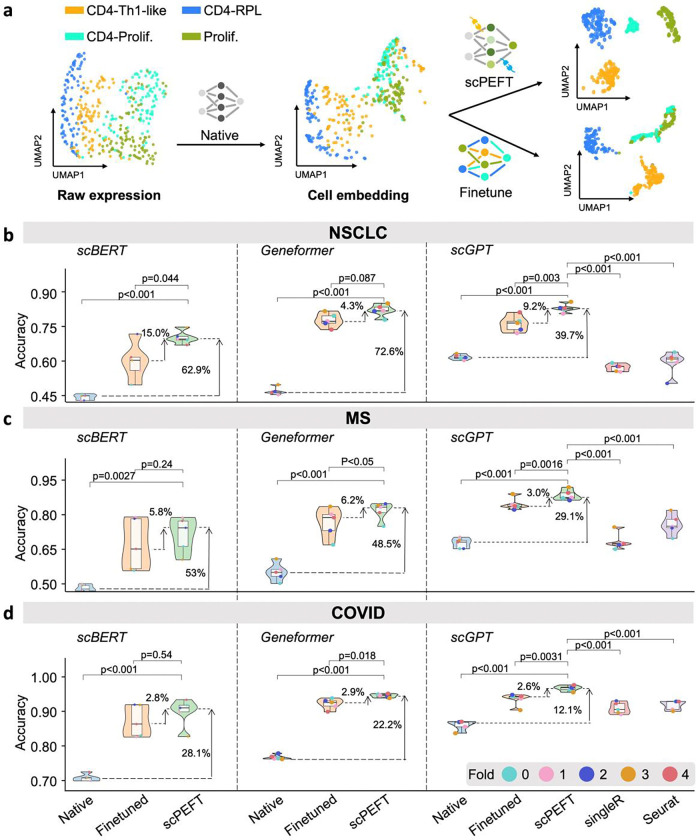
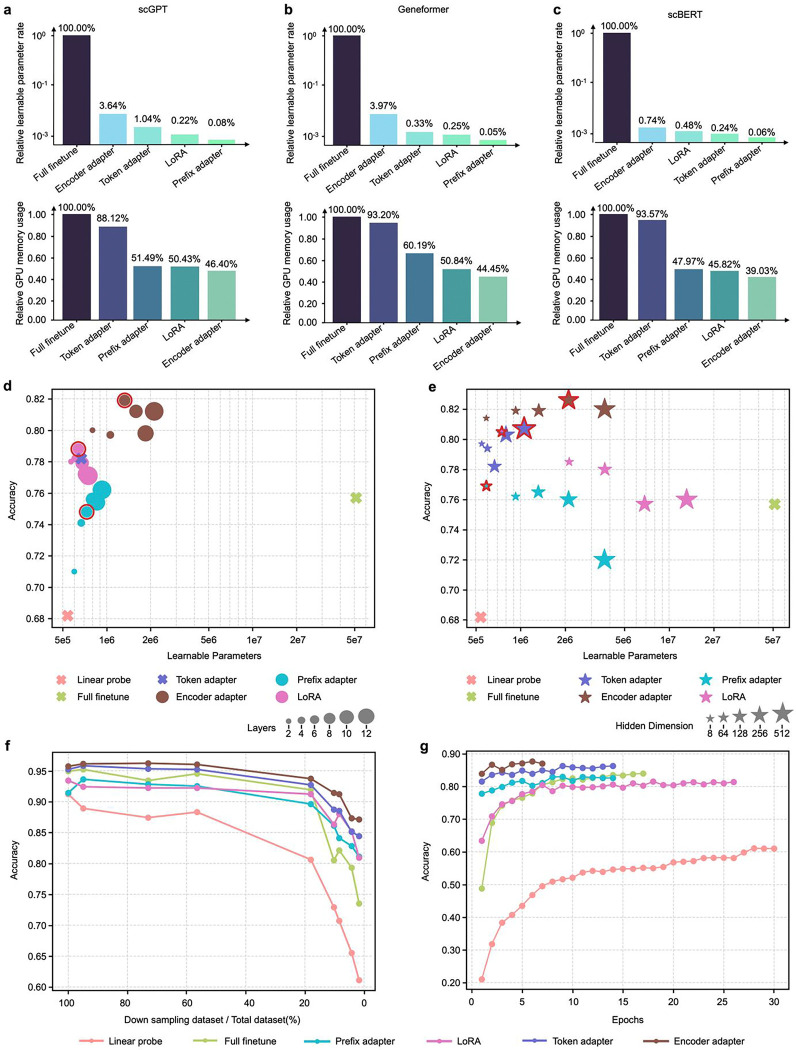



References
-
- Yang F, Wang W, Wang F, et al. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nature Machine Intelligence, 2022, 4(10): 852–866.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
