

Emergent glutamate & dopamine dysfunction in VPS35(D620N) knock-in mice and rapid reversal by LRRK2 inhibition
- PMID: 40319043
- PMCID: PMC12049453
- DOI: 10.1038/s41531-025-00948-7
Emergent glutamate & dopamine dysfunction in VPS35(D620N) knock-in mice and rapid reversal by LRRK2 inhibition
Abstract
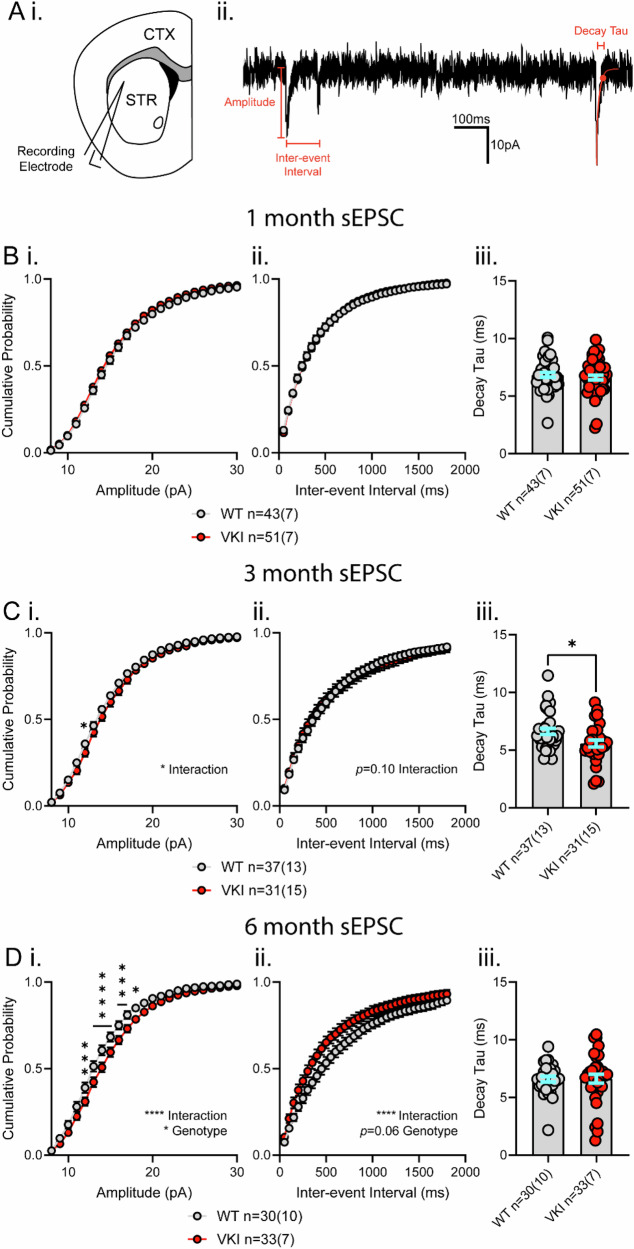
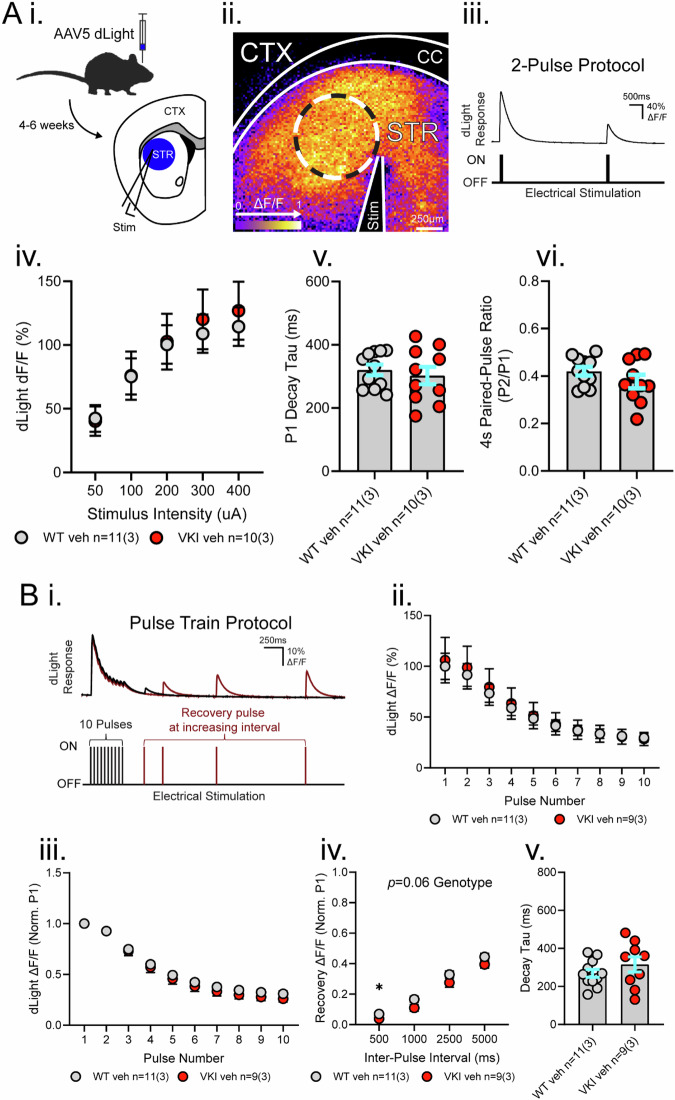
The D620N variant in Vacuolar Protein Sorting 35 (VPS35) causes autosomal-dominant, late-onset Parkinson's disease. VPS35 is a core subunit of the retromer complex that canonically recycles transmembrane cargo from sorting endosomes. Although retromer cargoes include many synaptic proteins, VPS35's neuronal functions are poorly understood. To investigate the consequences of the Parkinson's mutation, striatal neurotransmission was assessed in 1- to 6-month-old VPS35 D620N knock-in (VKI) mice. Spontaneous and optogenetically-evoked corticostriatal glutamate transmission was increased in VKI spiny projection neurons by 6 months and was unaffected by acute leucine-rich repeat kinase 2 (LRRK2) inhibition. Total striatal glutamate release by iGluSnFR imaging was similar to wild-type. dLight imaging revealed robust increases in VKI striatal dopamine release by 6 months, which were reversed with acute LRRK2 kinase inhibition. We conclude that increased striatal neurotransmission in VKI mice progressively emerges in young-adulthood, and that dopamine dysfunction is likely the result of sustained, rapidly-reversible, LRRK2 hyperactivity.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures





References
-
- Kim, H. J., Jeon, B. S. & Jenner, P. Hallmarks of Treatment Aspects: Parkinson’s Disease Throughout Centuries Including l -Dopa. in International Review of Neurobiology vol. 132 295–343 (Elsevier, 2017). - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
