

Huntingtin inclusion bodies have distinct immunophenotypes and ubiquitination profiles in the Huntington's disease human cerebral cortex
- PMID: 40319093
- PMCID: PMC12049496
- DOI: 10.1038/s41598-025-00465-w
Huntingtin inclusion bodies have distinct immunophenotypes and ubiquitination profiles in the Huntington's disease human cerebral cortex
Abstract
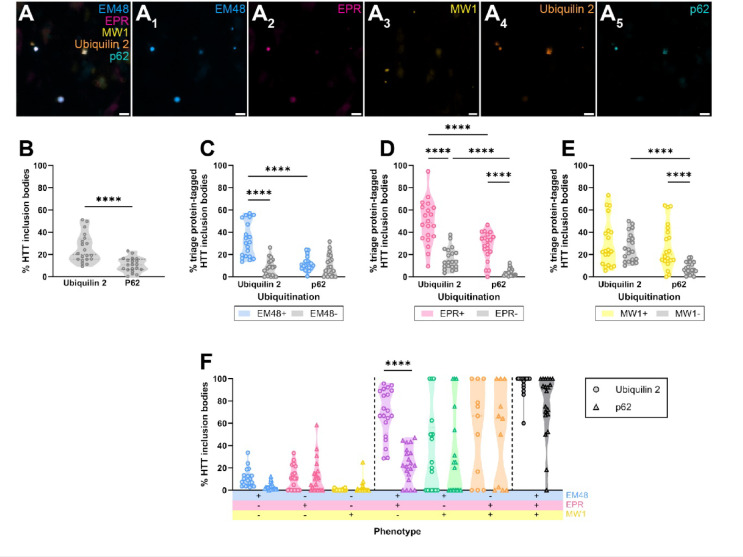
Huntington's disease (HD) is a hereditary neurodegenerative condition caused by a CAG repeat expansion mutation in the gene encoding the huntingtin (HTT) protein. The accumulation of HTT inclusion bodies is a pathological hallmark of HD and a common target for therapeutic strategies. However, the limited efficacy of treatments targeting the HTT protein highlights the need for a better understanding of the role of HTT inclusion bodies in HD pathogenesis. This study examined the heterogeneity of HTT inclusion body composition by co-labelling with three HTT epitope-specific antibodies to characterize HTT inclusion body 'immunophenotype'. We then characterized the size and sub-cellular location of HTT inclusions with distinct immunophenotypes. Using multiplex immunohistochemistry, we also examined the ubiquitination profile of each immunophenotype. Our findings demonstrate that HTT inclusions have a range of immunophenotypes, with some labelled by only one of the three antibodies and others exhibiting co-labelling by several antibodies, thus demonstrating the heterogeneity in inclusion composition and structure. We outline evidence that inclusion bodies exclusively labelled with the EM48 antibody are small, non-nuclear, and more abundant in HD cases with increased CAG repeat length, higher Vonsattel grade, and earlier age of onset. We also find that HTT inclusion bodies labelled by multiple antibodies are more likely to be ubiquitinated, predominantly by K63- rather than K48-linked ubiquitin, suggesting preferential degradation by autophagy. Lastly, we show that ubiquitinated HTT inclusion bodies are more highly immunoreactive for ubiquilin 2 than p62. Our findings highlight the need for multiple antibodies to capture the full spectrum of HTT pathology in HD and imply that future studies should consider the diversity of inclusion body composition and structure when correlating pathology formation to neurodegeneration, clinical symptoms, or disease severity.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests. Ethics approval and consent to participate: The tissue was donated with written informed consent from the donor and family before brain removal. All research procedures and protocols were approved by the Health and Disability Ethics Committee (Ref.14/NTA/208). Consent for publication: Written informed consent to publish anonymised data pertaining to tissue donors was obtained from next of kin at the time of tissue donation. All research procedures and protocols were approved by the Health and Disability Ethics Committee (Ref.14/NTA/208).
Figures





References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
