

Progress of personalized medicine of cystic fibrosis in the times of efficient CFTR modulators
- PMID: 40320452
- PMCID: PMC12050259
- DOI: 10.1186/s40348-025-00194-0
Progress of personalized medicine of cystic fibrosis in the times of efficient CFTR modulators
Abstract
Background: Cystic fibrosis (CF) is a systemic disorder of exocrine glands that is caused by mutations in the CFTR gene.
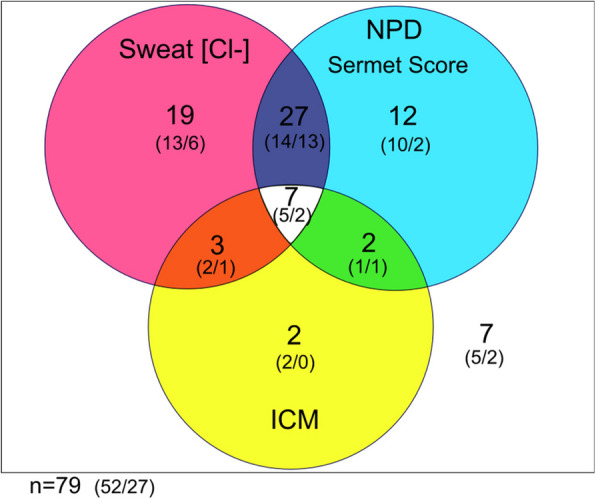
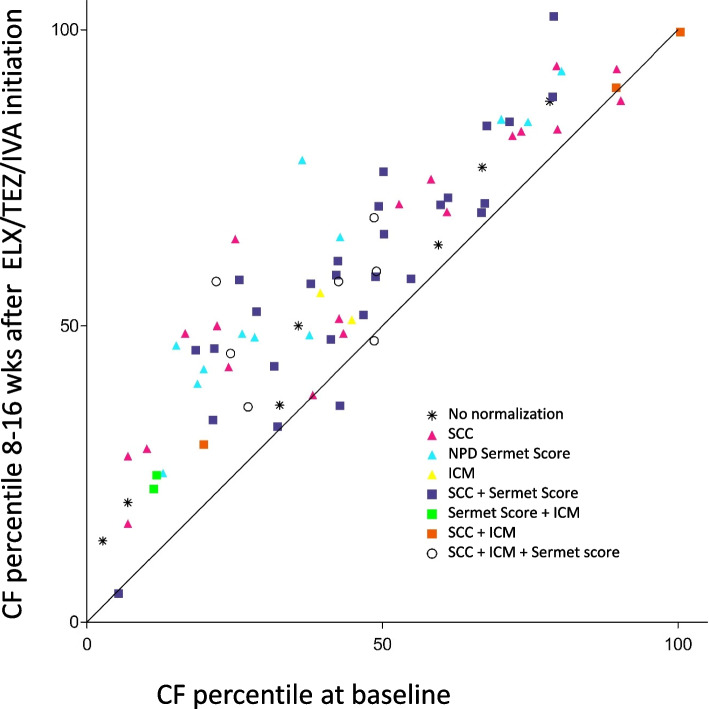
Main body: The basic defect in people with CF (pwCF) leads to impaired epithelial transport of chloride and bicarbonate that can be assessed by CFTR biomarkers, i.e. the β-adrenergic sweat rate and sweat chloride concentration (SCC), chloride conductance of the nasal respiratory epithelium (NPD), urine secretion of bicarbonate, intestinal current measurements (ICM) of chloride secretory responses in rectal biopsies and in bioassays of chloride transport in organoids or cell cultures. CFTR modulators are a novel class of drugs that improve defective posttranslational processing, trafficking and function of mutant CFTR. By April 2025, triple combination therapy with the CFTR potentiator ivacaftor (IVA) and the CFTR correctors elexacaftor (ELX) and tezacaftor (TEZ) has been approved in Europe for the treatment of all pwCF who do not carry two minimal function CFTR mutations. Previous phase 3 and post-approval phase 4 studies in pwCF who harbour one or two alleles of the major mutation F508del consistently reported significant improvements of lung function and anthropometry upon initiation of ELX/TEZ/IVA compared to baseline. Normalization of SCC, NPD and ICM correlated with clinical outcomes on the population level, but the restoration of CFTR function was diverse and not predictive for clinical outcome in the individual patient. Theratyping of non-F508del CF genotypes in patient-derived organoids and cell cultures revealed for most cases clinically meaningful increases of CFTR activity upon exposure to ELX/TEZ/IVA. Likewise, every second CF patient with non-F508del genotypes improved in SCC and clinical outcome upon exposure to ELX/TEZ/IVA indicating that triple CFTR modulator therapy is potentially beneficial for all pwCF who do not carry two minimal function CFTR mutations. This group who is not eligible for CFTR modulators may opt for gene addition therapy in the future, as the first-in-human trial with a recombinant lentiviral vector is underway.
Future directions: The upcoming generation of pwCF will probably experience a rather normal life in childhood and adolescence. To classify the upcoming personal signatures of CF disease in the times of efficient modulators, we need more sensitive CFTR biomarkers that address the long-term course of airway and gut microbiome, host defense, epithelial homeostasis and multiorgan metabolism.
Keywords: Biomarker; CFTR; Cystic fibrosis; Elexacaftor; Gene therapy; Ivacaftor; Tezacaftor; Theratyping.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: B.T. reports grants and contracts from the German Research Foundation (DFG), the German Federal Ministry of Education and Research (BMBF) and the Volkswagen Stiftung with payments made to the institution; personal fees for advisory board participation and educational events from Vertex Pharmaceuticals; lecture honoraria from Vertex Pharmaceuticals and streamedup! GmbH. S.T.P. reports grants and contracts from the Mukoviszidose e.V., the Else Kröner Fresenius Foundation and the Deutsche Forschungsgemeinschaft (DFG) with payments made to the institution and travel support from Vertex Pharmaceuticals. A-M.D. reports grants and contracts from the German Federal Ministry of Education and Research (BMBF), Vertex Pharmaceuticals Incorporated, European Cystic Fibrosis Society Clinical Trial Network (ECFS-CTN) and Christiane Herzog Stiftung with payments made to the institution, personal fees from the c4c consortium, GSK and European Cystic Fibrosis Society. S.Y.G. reports grants from Mukoviszidose e.V. (German CF Foundation) and Vertex Pharmaceuticals Incorporated with payments made to institution; personal fees for advisory board participation from Chiesi GmbH and Vertex Pharmaceuticals Incorporated; lecture honoraria and honoraria for a CME module from Vertex Pharmaceuticals Incorporated. L.N. reports grants and contracts from the German Federal Ministry of Education and Research (BMBF), the European Cystic Fibrosis Society and Vertex Pharmaceuticals with payments made to the institution. O. S. reports grants from the German Federal Ministry of Education and Research (BMBF) with payment made to the institution and honoraria from Vertex Pharmaceuticals Incorporated for lectures. M.A.M. reports grants and contracts from the German Research Foundation (DFG), the German Federal Ministry of Education and Research (BMBF), Boehringer Ingelheim, Enterprise Therapeutics and Vertex Pharmaceuticals with payments made to the institution; personal fees for advisory board participation or consulting from Boehringer Ingelheim, Enterprise Therapeutics, Kither Biotech, Splisense, Vertex Pharmaceuticals; lecture honoraria from Vertex Pharmaceuticals; and travel support from Boehringer Ingelheim and Vertex Pharmaceuticals.
Figures
References
-
- Childs B (1999) Genetic Medicine: A Logic of Disease. Johns Hopkins University Press, Baltimore
-
- Mall MA, Burgel PR, Castellani C, Davies JC, Salathe M, Taylor-Cousar JL (2024) Cystic fibrosis. Nat Rev Dis Primers 10:53. 10.1038/s41572-024-00538-6 - PubMed
-
- Grasemann H, Ratjen F (2023) Cystic Fibrosis. N Engl J Med 389:1693–1707. 10.1056/NEJMra2216474 - PubMed
-
- Guo J, King I, Hill A (2024) International disparities in diagnosis and treatment access for cystic fibrosis. Pediatr Pulmonol 59:1622–1630. 10.1002/ppul.26954 - PubMed
-
- Bobadilla JL, Macek M Jr, Fine JP, Farrell PM (2002) Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum Mutat 19:575–606. 10.1002/humu.10041 - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
