

The anti-diabetic PPARγ agonist Pioglitazone inhibits cell proliferation and induces metabolic reprogramming in prostate cancer
- PMID: 40320521
- PMCID: PMC12051277
- DOI: 10.1186/s12943-025-02320-y
The anti-diabetic PPARγ agonist Pioglitazone inhibits cell proliferation and induces metabolic reprogramming in prostate cancer
Abstract
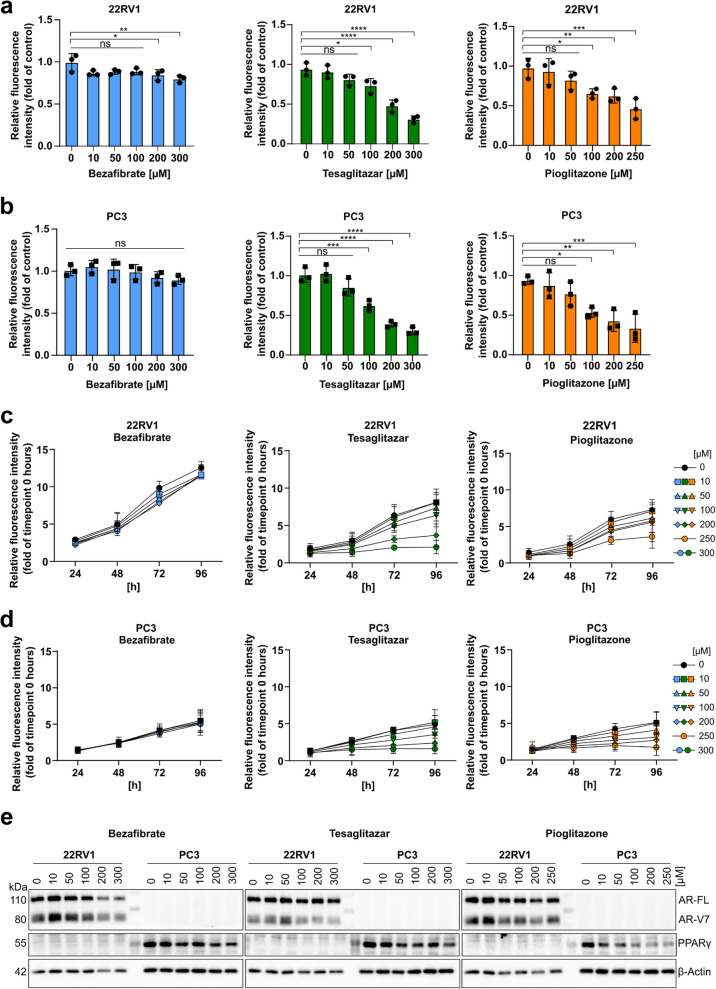
Prostate cancer (PCa) and Type 2 diabetes (T2D) often co-occur, yet their relationship remains elusive. While some studies suggest that T2D lowers PCa risk, others report conflicting data. This study investigates the effects of peroxisome proliferator-activated receptor (PPAR) agonists Bezafibrate, Tesaglitazar, and Pioglitazone on PCa tumorigenesis. Analysis of patient datasets revealed that high PPARG expression correlates with advanced PCa and poor survival. The PPARγ agonists Pioglitazone and Tesaglitazar notably reduced cell proliferation and PPARγ protein levels in primary and metastatic PCa-derived cells. Proteomic analysis identified intrinsic differences in mTORC1 and mitochondrial fatty acid oxidation (FAO) pathways between primary and metastatic PCa cells, which were further disrupted by Tesaglitazar and Pioglitazone. Moreover, metabolomics, Seahorse Assay-based metabolic profiling, and radiotracer uptake assays revealed that Pioglitazone shifted primary PCa cells' metabolism towards glycolysis and increased FAO in metastatic cells, reducing mitochondrial ATP production. Furthermore, Pioglitazone suppressed cell migration in primary and metastatic PCa cells and induced the epithelial marker E-Cadherin in primary PCa cells. In vivo, Pioglitazone reduced tumor growth in a metastatic PC3 xenograft model, increased phosho AMPKα and decreased phospho mTOR levels. In addition, diabetic PCa patients treated with PPAR agonists post-radical prostatectomy implied no biochemical recurrence over five to ten years compared to non-diabetic PCa patients. Our findings suggest that Pioglitazone reduces PCa cell proliferation and induces metabolic and epithelial changes, highlighting the potential of repurposing metabolic drugs for PCa therapy.
Keywords: Cancer therapy; Energy metabolism; Extracellular acidification; Metabolic rewiring; Oxygen consumption rate; PPAR agonists; Type 2 diabetes mellitus (T2DM).
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethical Approval and consent to participate: The retrospective data analysis was approved by the local ethics committee of Medical University Innsbruck (Vote number: 1301/2023) and performed according to the Declaration of Helsinki. The patient's consent for the use of residual material was obtained. All animal procedures were carried out under UK Home Office license PP0918061 according to the Animals (Scientific Procedures) Act 1986 and were approved by the University of Cambridge Animal Welfare and Ethical Review Board (AWERB). Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures






References
-
- Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
