

Differential effects of tofacitinib on macrophage activation contribute to lack of response in ulcerative colitis patients
- PMID: 40320713
- PMCID: PMC12137895
- DOI: 10.1093/ecco-jcc/jjaf076
Differential effects of tofacitinib on macrophage activation contribute to lack of response in ulcerative colitis patients
Abstract
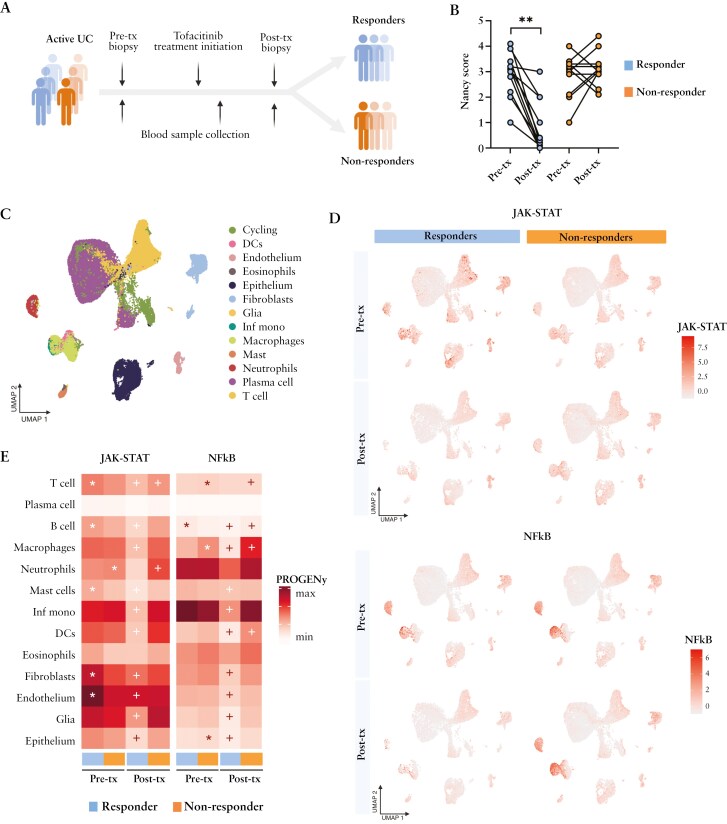
Background and aims: Tofacitinib, a Janus kinase inhibitor, is approved for the treatment of moderate-to-severe ulcerative colitis. Nonetheless, 40-60% of patients will not respond adequately. The mechanisms underlying responses to tofacitinib remain unknown.
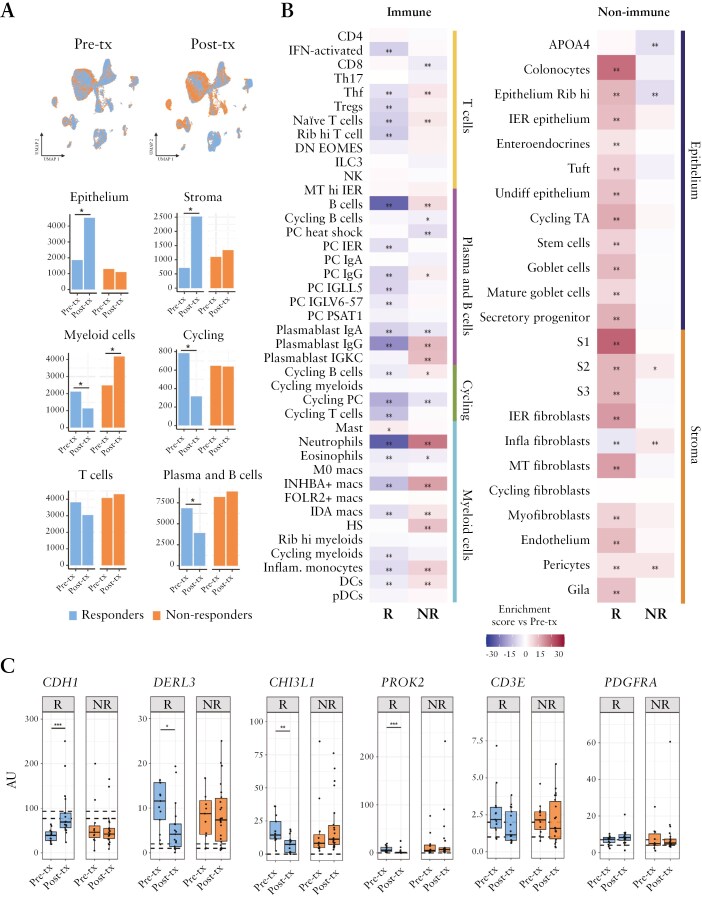
Methods: We applied single-cell and/or bulk RNA analysis to biopsies (n = 23 and 63, respectively) from ulcerative colitis patients (n = 31) before and after tofacitinib treatment. Response was assessed using endoscopic and clinical criteria. In vitro-derived macrophages and primary intestinal fibroblasts were used to validate our findings.
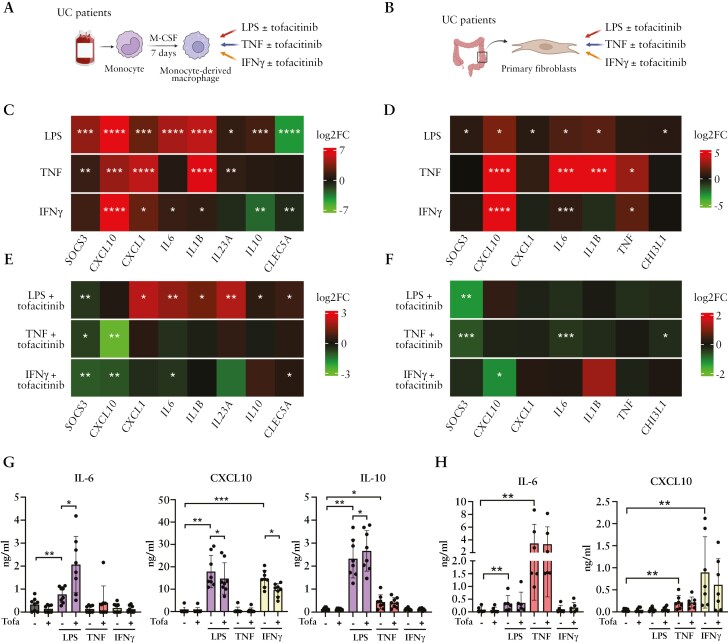
Results: Forty percent of patients responded to tofacitinib. Responders exhibited higher baseline JAK-STAT activity, while non-responders had increased baseline NF-kB pathway activation. Response was associated with significant changes in the abundance and/or activation of immune, epithelial, and stromal cells and the downregulation of S100A9, FCGR3A, MMP12 in resident macrophages. In contrast, non-responders showed a significant increase in the number and activation of macrophages and fibroblasts following tofacitinib treatment, including upregulation of MMP9, IL1B, IL6, CXCL1, CXCL8, and S100A9 compared to baseline. In monocyte-derived macrophages tofacitinib drove the hyperactivation of macrophages in response to lipopolysaccharide, but not TNF or IFNγ. This effect is dependent on the inhibition of IL-10 signaling, which is abundantly induced in response to LPS, but not to TNF or IFNγ. In contrast, cultured fibroblasts, which produced no IL-10 regardless of the stimuli, showed no hyperactivation when pre-treated with tofacitinib.
Conclusions: We conclude that resistance to tofacitinib is mediated by the hyperactivation of myeloid cells and we identify IL-10-dependent macrophages as one cellular subset contributing to this resistance.
Keywords: macrophage; single-cell RNA sequencing; tofacitinib.
© The Author(s) 2025. Published by Oxford University Press on behalf of European Crohn’s and Colitis Organisation.
Conflict of interest statement
Figures



References
-
- Mereu E, Lafzi A, Moutinho C, et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat Biotechnol. 2020;38:747–755. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
