

Redox and actin, a fascinating story
- PMID: 40328105
- PMCID: PMC12127579
- DOI: 10.1016/j.redox.2025.103630
Redox and actin, a fascinating story
Abstract
Actin is an extraordinarily complex protein whose functions are essential to cell motility, division, contraction, signaling, transport, tissular structures, DNA repair, and many more cellular activities critical to life for both animals and plants. It is one of the most abundant and conserved proteins and it exists in either a soluble, globular (monomeric, G-actin) or an insoluble, self-assembled (polymerized or filamentous actin, F-actin) conformation as a key component of the cytoskeleton. In the early 1990's little, if anything, was known about the impact of reactive oxygen species (ROS) on the biology of actin except that ROS could disrupt the actin cytoskeleton. Instructively, G-actin is susceptible to alteration by ROS, and thus, purification of G-actin is typically performed in the presence of strong antioxidants (like dithiothreitol) to limit its oxidative degradation. In contrast, F-actin is a more stable conformation and thus actin can be kept relatively intact in purified preparations as filaments at low temperature for extended periods of time. Both G- and F-actin interact with a myriad of intracellular proteins and at least with a couple of extracellular proteins, and these interactions are essential to the many actin functions. This review will show how, over the past 30 years, our understanding of the role of ROS for actin biology has evolved from noxious denaturizing agents to remarkable regulators of the actin cytoskeleton in cells and consequent cellular functions.
Keywords: ADF/Cofilin; Actin; Atherosclerosis; Bubbles; Cancer; Cell motility; Cytoskeleton; EGF-Receptor; Hydrogen peroxide; Inflammation; Kaposi's sarcoma; Lamellipodium; MICAL; Membranes; NADPH-Oxidase; NOX-1; NOX-2; PDGF-Receptor; Phosphatases; Phosphatidylinositol 4,5 bisphosphate; Phospholipase C-y1; Profilin; Rac1; Ras; Reactive oxygen species; Regeneration; Superoxide; Tissue repair.
Copyright © 2025 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors do not have a conflict of interest.
Figures
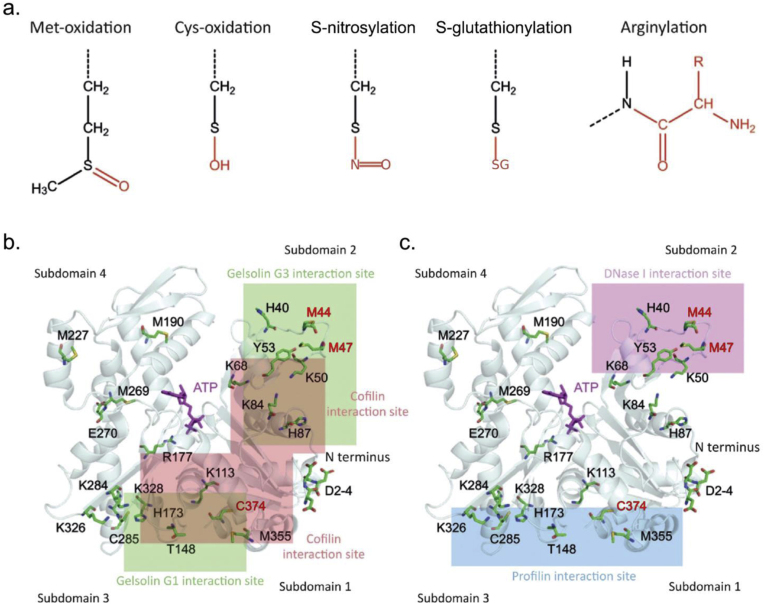
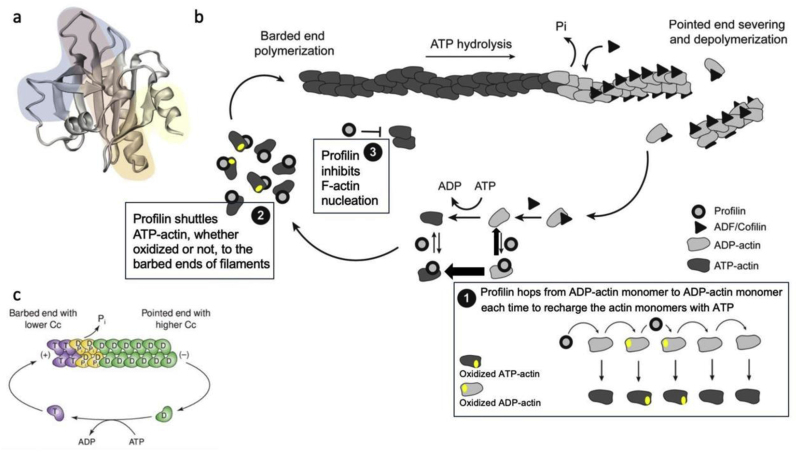
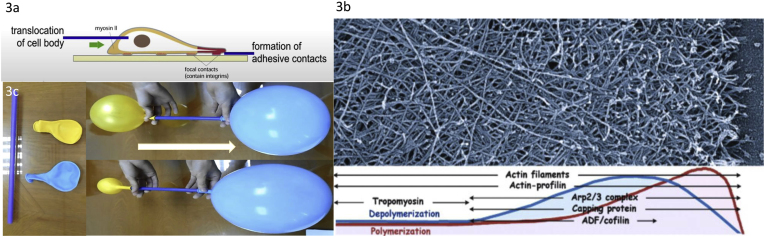
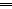 C/r where P is the pressure inside a bubble, C in cells is a constant, and r is the radius of the bubble [35]. Hence, the smaller the bubble the greater the pressure inside the bubble. This continuously advancing actin compression machinery results in pressurizing the plasma membrane forward while stabilizing each advance through forward treadmilling and branching of actin filaments. Equilibrium of pressure across the cell is maintained by the cytoskeleton (possibly involving the wrinkles) [31] and the myosin mediated contraction at the rear end of the cell which detaches from the substrate as the cell advances. When two balloons of identical initial size when deflated, are inflated with one less than the other, upon connecting the balloons with a straw, the transfer of air is from the smaller balloon to the larger balloon, in a way that illustrates the bubble experiments of Laplace. (d) Molecular activities at the edge of the lamellipodia that support the compression machinery (figure adapted from the article of Thomas Pollard and Gary Borisy) [3]. Reorganization of the actin cytoskeleton involves the metabolism of membrane phosphatidylinositols, followed by activation of Rac1 through interaction with guanine nucleotide exchange factor Tiam-1 which is bound to phosphatidylinositol 4,5 bisphosphate and accelerates the binding of Rac1 to GTP. In turn, GTP-Rac1 activates NADPH oxidase (NOX1) with production of superoxide and ROS including H2O2 (green gradient in Fig. 3d), which contributes to the activation of WASP/Scar proteins. WASP/Scar bring together the Arp2/3 complex that nucleates new filaments and to form new branches of actin filaments at the edge of the lamellipodia. Such branching of filaments are necessary to generate the compression machinery of the lamellipodia at the edge. Capping proteins can limit the length of filaments, and filaments age by hydrolysis of ATP bound to each actin subunit of filaments (light blue to dark blue) followed by dissociation of the phosphate to complete the ATPase cycle of actin (see Fig. 3). ADF/cofilin promotes phosphate dissociation, severs ADP-actin filaments and promotes dissociation of ADP-actin from filament pointed ends. MICAL oxidation of actin filaments can accelerate markedly the depolymerization of filaments produced by ADP/cofilin. Profilin catalyzes the exchange of ADP for ATP (turning the subunits light blue again), thus returning actin subunits to the pool of ATP-actin bound to profilin, ready to elongate barbed ends as they become available. For MICAL oxidized G-actin, an additional step of reduction catalyzed by methionine sulfoxide reductase B1 (MsrB1) is required for G-actin to join the pool of ATP-actin bound to profilin and ready for polymerization.
C/r where P is the pressure inside a bubble, C in cells is a constant, and r is the radius of the bubble [35]. Hence, the smaller the bubble the greater the pressure inside the bubble. This continuously advancing actin compression machinery results in pressurizing the plasma membrane forward while stabilizing each advance through forward treadmilling and branching of actin filaments. Equilibrium of pressure across the cell is maintained by the cytoskeleton (possibly involving the wrinkles) [31] and the myosin mediated contraction at the rear end of the cell which detaches from the substrate as the cell advances. When two balloons of identical initial size when deflated, are inflated with one less than the other, upon connecting the balloons with a straw, the transfer of air is from the smaller balloon to the larger balloon, in a way that illustrates the bubble experiments of Laplace. (d) Molecular activities at the edge of the lamellipodia that support the compression machinery (figure adapted from the article of Thomas Pollard and Gary Borisy) [3]. Reorganization of the actin cytoskeleton involves the metabolism of membrane phosphatidylinositols, followed by activation of Rac1 through interaction with guanine nucleotide exchange factor Tiam-1 which is bound to phosphatidylinositol 4,5 bisphosphate and accelerates the binding of Rac1 to GTP. In turn, GTP-Rac1 activates NADPH oxidase (NOX1) with production of superoxide and ROS including H2O2 (green gradient in Fig. 3d), which contributes to the activation of WASP/Scar proteins. WASP/Scar bring together the Arp2/3 complex that nucleates new filaments and to form new branches of actin filaments at the edge of the lamellipodia. Such branching of filaments are necessary to generate the compression machinery of the lamellipodia at the edge. Capping proteins can limit the length of filaments, and filaments age by hydrolysis of ATP bound to each actin subunit of filaments (light blue to dark blue) followed by dissociation of the phosphate to complete the ATPase cycle of actin (see Fig. 3). ADF/cofilin promotes phosphate dissociation, severs ADP-actin filaments and promotes dissociation of ADP-actin from filament pointed ends. MICAL oxidation of actin filaments can accelerate markedly the depolymerization of filaments produced by ADP/cofilin. Profilin catalyzes the exchange of ADP for ATP (turning the subunits light blue again), thus returning actin subunits to the pool of ATP-actin bound to profilin, ready to elongate barbed ends as they become available. For MICAL oxidized G-actin, an additional step of reduction catalyzed by methionine sulfoxide reductase B1 (MsrB1) is required for G-actin to join the pool of ATP-actin bound to profilin and ready for polymerization.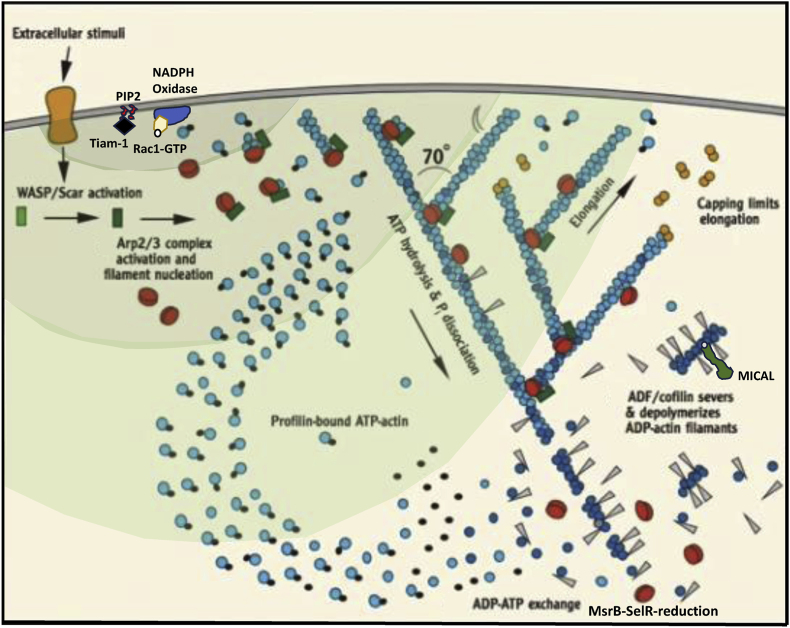 C/r where P is the pressure inside a bubble, C in cells is a constant, and r is the radius of the bubble [35]. Hence, the smaller the bubble the greater the pressure inside the bubble. This continuously advancing actin compression machinery results in pressurizing the plasma membrane forward while stabilizing each advance through forward treadmilling and branching of actin filaments. Equilibrium of pressure across the cell is maintained by the cytoskeleton (possibly involving the wrinkles) [31] and the myosin mediated contraction at the rear end of the cell which detaches from the substrate as the cell advances. When two balloons of identical initial size when deflated, are inflated with one less than the other, upon connecting the balloons with a straw, the transfer of air is from the smaller balloon to the larger balloon, in a way that illustrates the bubble experiments of Laplace. (d) Molecular activities at the edge of the lamellipodia that support the compression machinery (figure adapted from the article of Thomas Pollard and Gary Borisy) [3]. Reorganization of the actin cytoskeleton involves the metabolism of membrane phosphatidylinositols, followed by activation of Rac1 through interaction with guanine nucleotide exchange factor Tiam-1 which is bound to phosphatidylinositol 4,5 bisphosphate and accelerates the binding of Rac1 to GTP. In turn, GTP-Rac1 activates NADPH oxidase (NOX1) with production of superoxide and ROS including H2O2 (green gradient in Fig. 3d), which contributes to the activation of WASP/Scar proteins. WASP/Scar bring together the Arp2/3 complex that nucleates new filaments and to form new branches of actin filaments at the edge of the lamellipodia. Such branching of filaments are necessary to generate the compression machinery of the lamellipodia at the edge. Capping proteins can limit the length of filaments, and filaments age by hydrolysis of ATP bound to each actin subunit of filaments (light blue to dark blue) followed by dissociation of the phosphate to complete the ATPase cycle of actin (see Fig. 3). ADF/cofilin promotes phosphate dissociation, severs ADP-actin filaments and promotes dissociation of ADP-actin from filament pointed ends. MICAL oxidation of actin filaments can accelerate markedly the depolymerization of filaments produced by ADP/cofilin. Profilin catalyzes the exchange of ADP for ATP (turning the subunits light blue again), thus returning actin subunits to the pool of ATP-actin bound to profilin, ready to elongate barbed ends as they become available. For MICAL oxidized G-actin, an additional step of reduction catalyzed by methionine sulfoxide reductase B1 (MsrB1) is required for G-actin to join the pool of ATP-actin bound to profilin and ready for polymerization.
C/r where P is the pressure inside a bubble, C in cells is a constant, and r is the radius of the bubble [35]. Hence, the smaller the bubble the greater the pressure inside the bubble. This continuously advancing actin compression machinery results in pressurizing the plasma membrane forward while stabilizing each advance through forward treadmilling and branching of actin filaments. Equilibrium of pressure across the cell is maintained by the cytoskeleton (possibly involving the wrinkles) [31] and the myosin mediated contraction at the rear end of the cell which detaches from the substrate as the cell advances. When two balloons of identical initial size when deflated, are inflated with one less than the other, upon connecting the balloons with a straw, the transfer of air is from the smaller balloon to the larger balloon, in a way that illustrates the bubble experiments of Laplace. (d) Molecular activities at the edge of the lamellipodia that support the compression machinery (figure adapted from the article of Thomas Pollard and Gary Borisy) [3]. Reorganization of the actin cytoskeleton involves the metabolism of membrane phosphatidylinositols, followed by activation of Rac1 through interaction with guanine nucleotide exchange factor Tiam-1 which is bound to phosphatidylinositol 4,5 bisphosphate and accelerates the binding of Rac1 to GTP. In turn, GTP-Rac1 activates NADPH oxidase (NOX1) with production of superoxide and ROS including H2O2 (green gradient in Fig. 3d), which contributes to the activation of WASP/Scar proteins. WASP/Scar bring together the Arp2/3 complex that nucleates new filaments and to form new branches of actin filaments at the edge of the lamellipodia. Such branching of filaments are necessary to generate the compression machinery of the lamellipodia at the edge. Capping proteins can limit the length of filaments, and filaments age by hydrolysis of ATP bound to each actin subunit of filaments (light blue to dark blue) followed by dissociation of the phosphate to complete the ATPase cycle of actin (see Fig. 3). ADF/cofilin promotes phosphate dissociation, severs ADP-actin filaments and promotes dissociation of ADP-actin from filament pointed ends. MICAL oxidation of actin filaments can accelerate markedly the depolymerization of filaments produced by ADP/cofilin. Profilin catalyzes the exchange of ADP for ATP (turning the subunits light blue again), thus returning actin subunits to the pool of ATP-actin bound to profilin, ready to elongate barbed ends as they become available. For MICAL oxidized G-actin, an additional step of reduction catalyzed by methionine sulfoxide reductase B1 (MsrB1) is required for G-actin to join the pool of ATP-actin bound to profilin and ready for polymerization.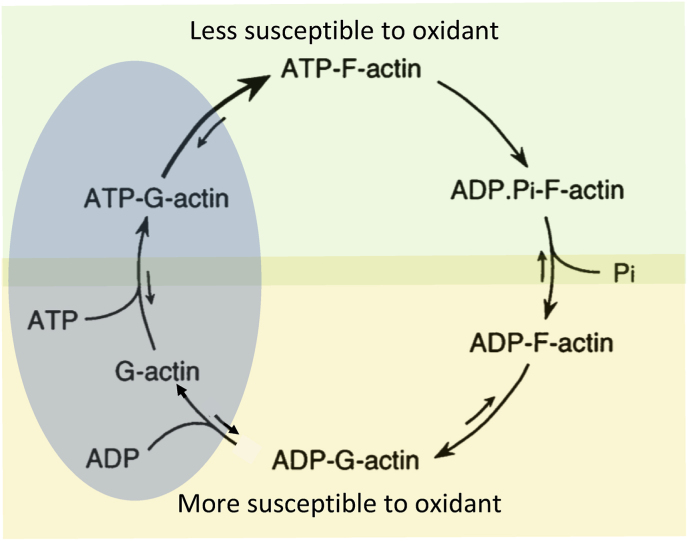
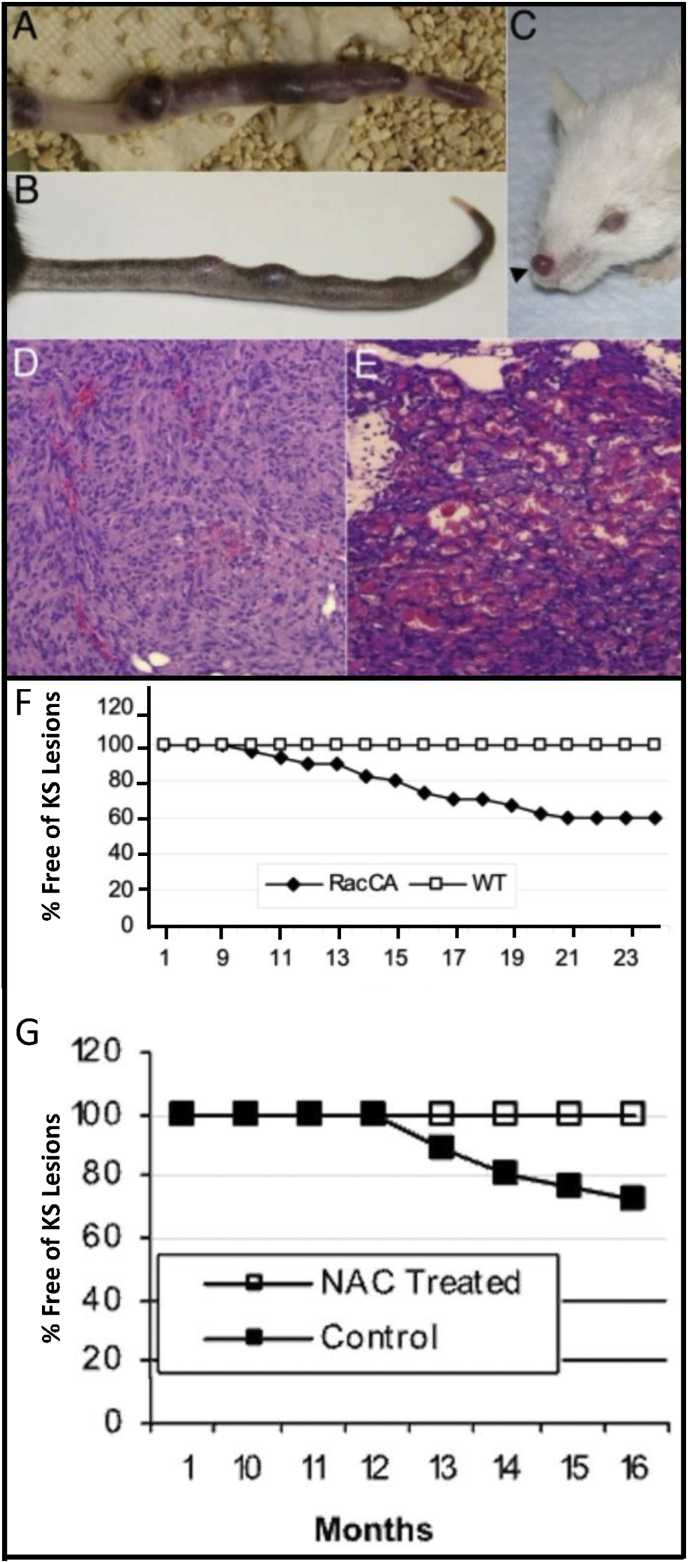
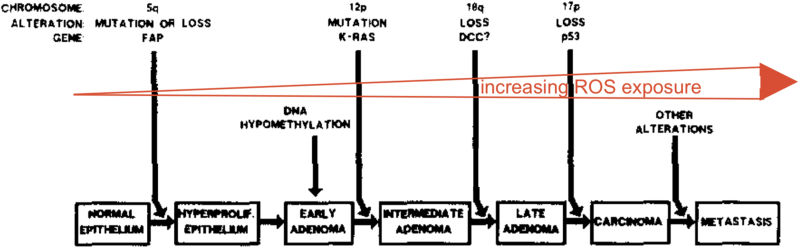
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
