

Trans-eQTL hotspots shape complex traits by modulating cellular states
- PMID: 40328252
- PMCID: PMC12143327
- DOI: 10.1016/j.xgen.2025.100873
Trans-eQTL hotspots shape complex traits by modulating cellular states
Abstract
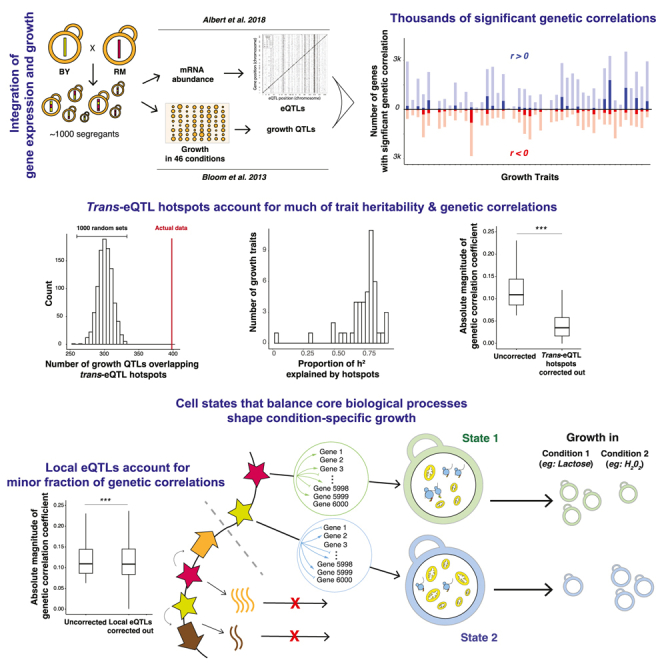
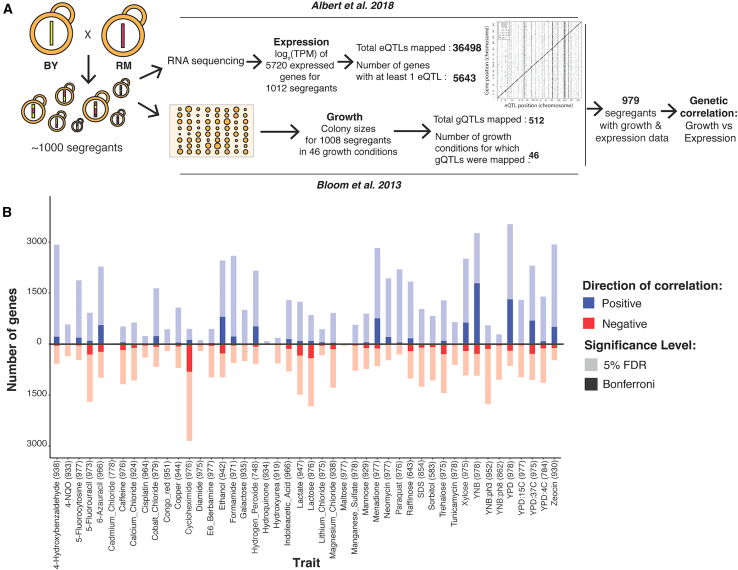
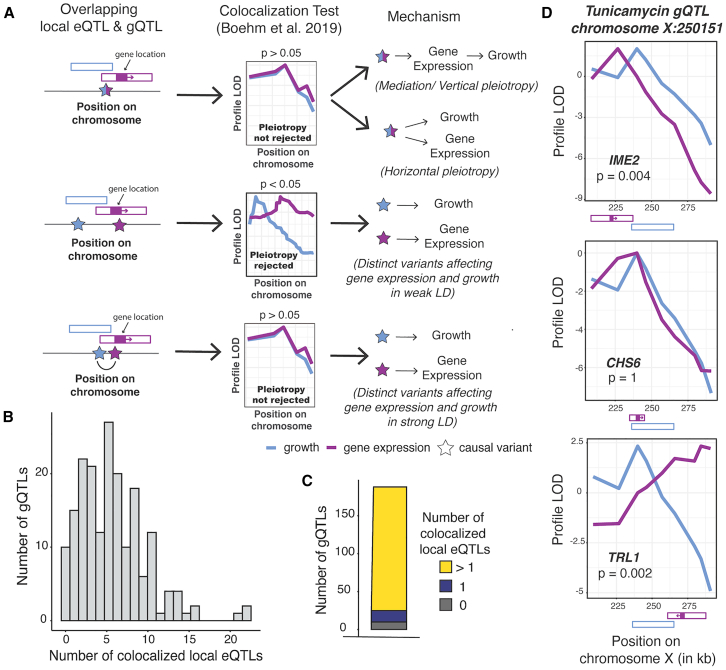
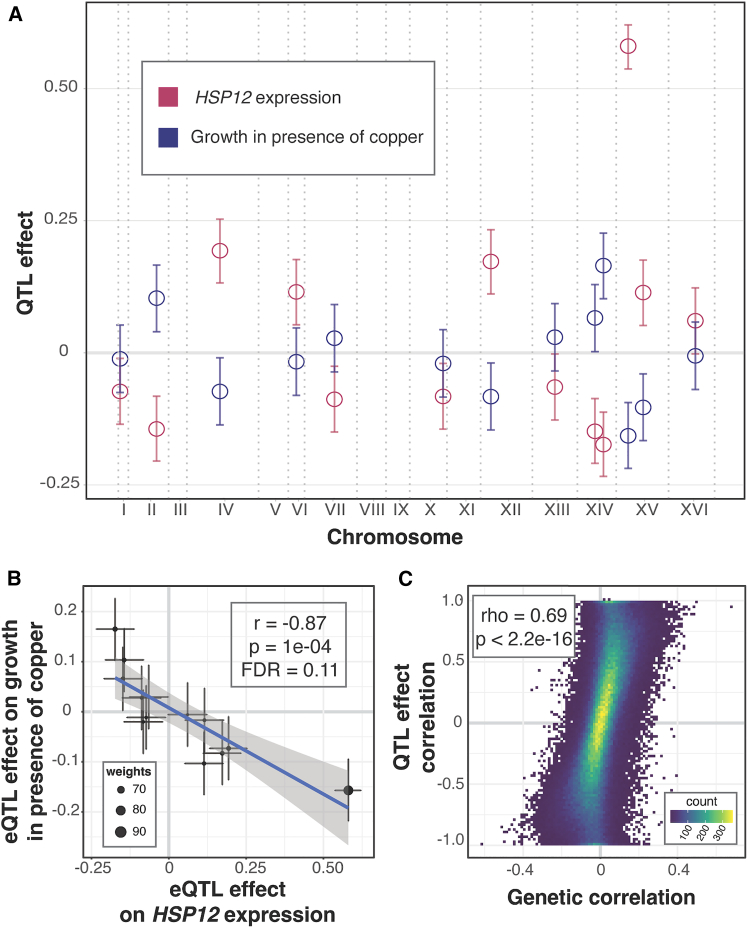
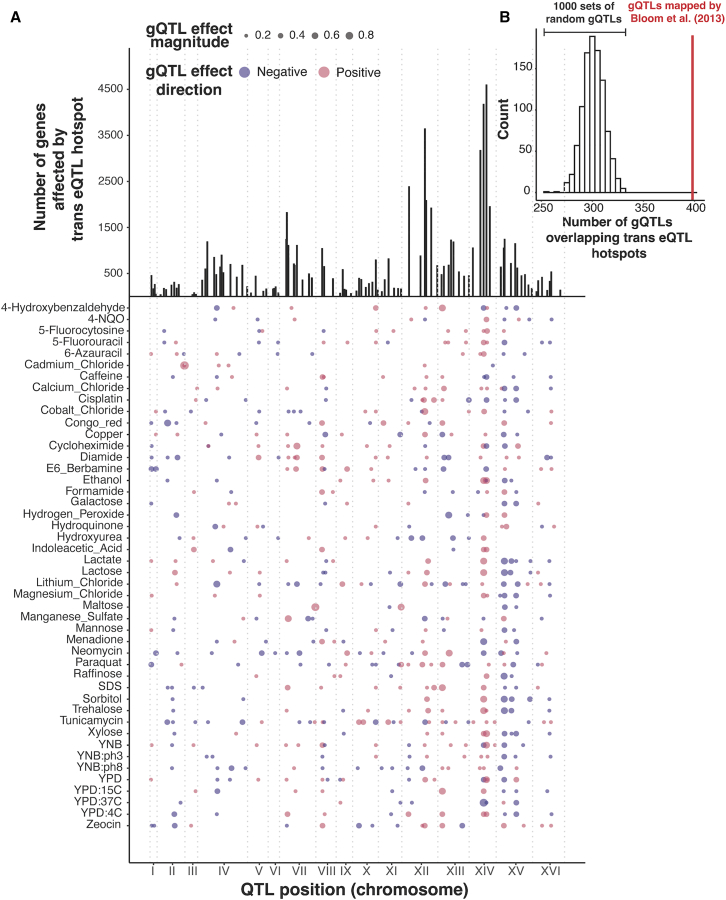
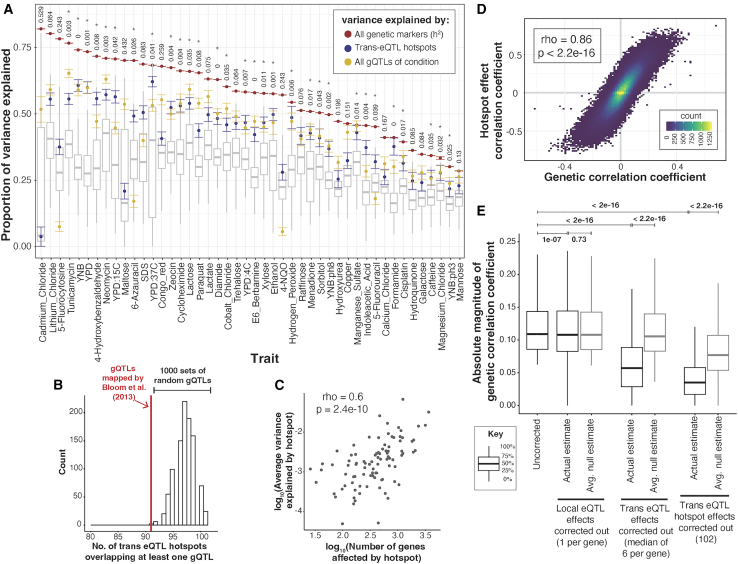
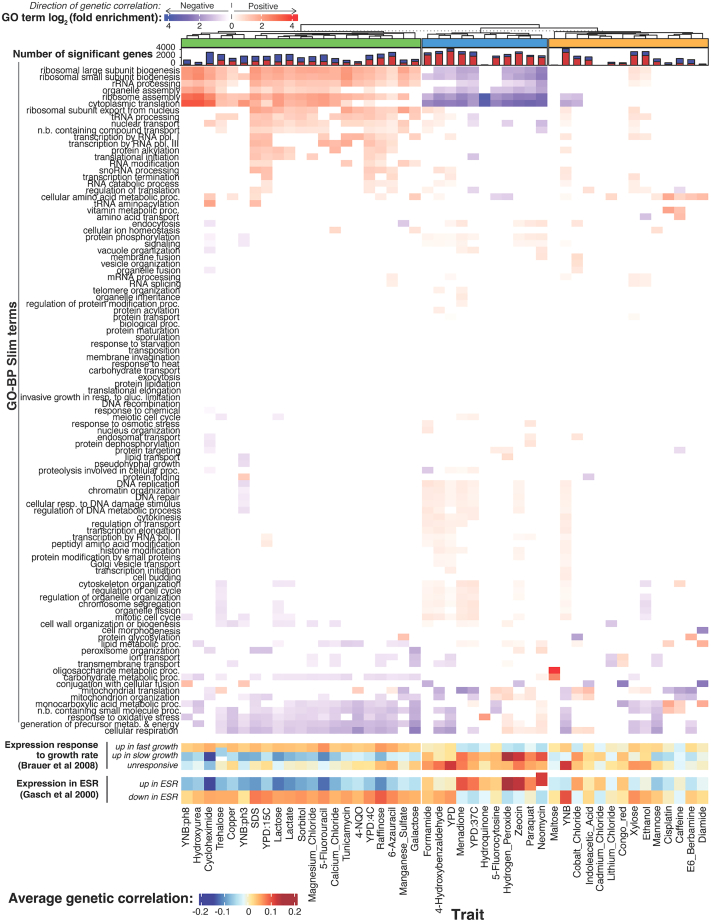
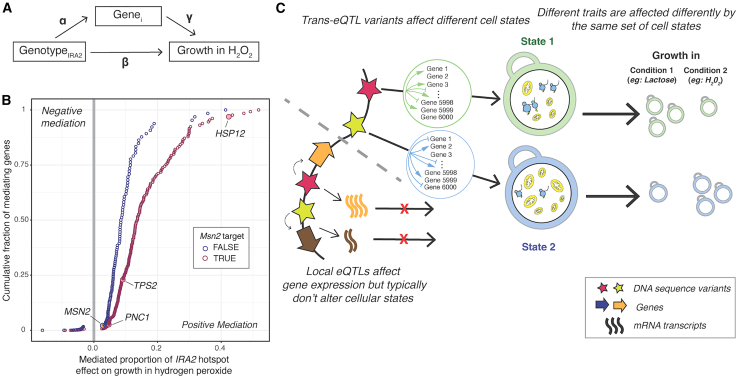
Regulatory genetic variation shapes gene expression, providing an important mechanism connecting DNA variation and complex traits. The causal relationships between gene expression and complex traits remain poorly understood. Here, we integrated transcriptomes and 46 genetically complex growth traits in a large cross between two strains of the yeast Saccharomyces cerevisiae. We discovered thousands of genetic correlations between gene expression and growth, suggesting potential functional connections. Local regulatory variation was a minor source of these genetic correlations. Instead, genetic correlations tended to arise from multiple independent trans-acting regulatory loci. Trans-acting hotspots that affect the expression of numerous genes accounted for particularly large fractions of genetic growth variation and of genetic correlations between gene expression and growth. Genes with genetic correlations were enriched for similar biological processes across traits but with heterogeneous direction of effect. Our results reveal how trans-acting regulatory hotspots shape complex traits by altering cellular states.
Keywords: QTLs; complex traits; expression QTLs; gene expression; genetic variation; heritability; mediation; pleiotropy; quantitative genetics; yeast.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Update of
-
Trans-eQTL hotspots shape complex traits by modulating cellular states.bioRxiv [Preprint]. 2024 Jul 8:2023.11.14.567054. doi: 10.1101/2023.11.14.567054. bioRxiv. 2024. Update in: Cell Genom. 2025 May 14;5(5):100873. doi: 10.1016/j.xgen.2025.100873. PMID: 38014174 Free PMC article. Updated. Preprint.
References
-
- Cusanovich D.A., Billstrand C., Zhou X., Chavarria C., De Leon S., Michelini K., Pai A.A., Ober C., Gilad Y. The combination of a genome-wide association study of lymphocyte count and analysis of gene expression data reveals novel asthma candidate genes. Hum. Mol. Genet. 2012;21:2111–2123. doi: 10.1093/hmg/dds021. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
