

Distinct methylomic signatures of high-altitude acclimatization and adaptation in the Tibetan Plateau
- PMID: 40328746
- PMCID: PMC12056056
- DOI: 10.1038/s41421-025-00795-z
Distinct methylomic signatures of high-altitude acclimatization and adaptation in the Tibetan Plateau
Abstract
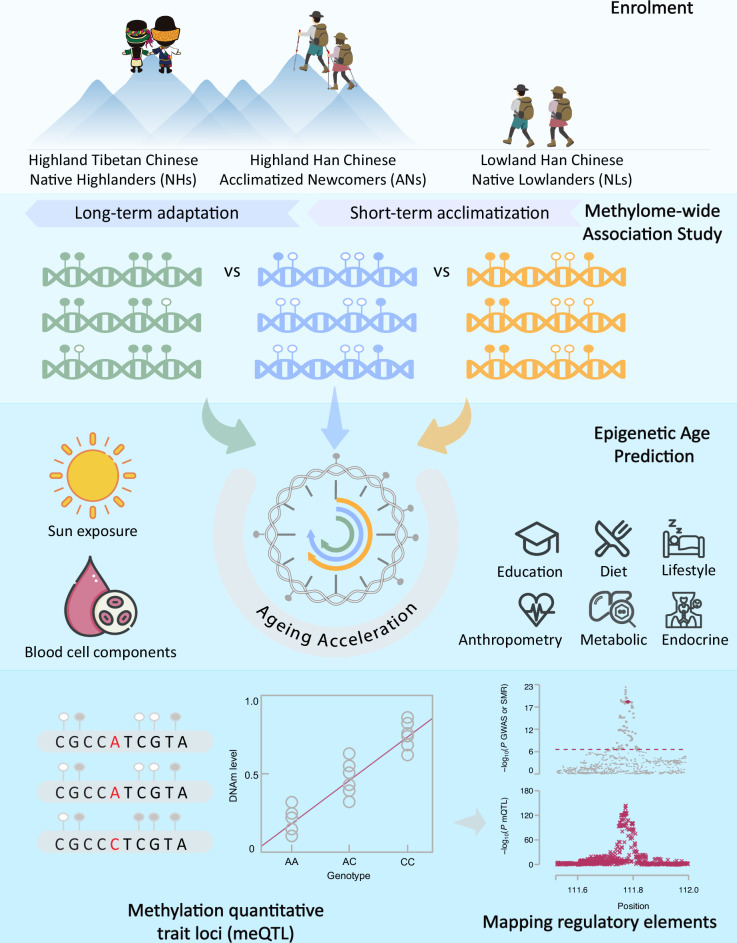
High altitude presents a challenging environment for human settlement. DNA methylation is an essential epigenetic mechanism that responds to environmental stimuli, but its roles in high-altitude short-term acclimatization (STA) and long-term adaptation (LTA) are poorly understood. Here, we conducted a methylome-wide association study involving 687 native highlanders and 299 acclimatized newcomers in the Tibetan Plateau and 462 native lowlanders to identify differentially methylated sites (DMSs) associated with STA or LTA. We identified 93 and 4070 DMSs for STA and LTA, respectively, which had no overlap, showed opposite asymmetric effect size patterns, and resided near genes enriched in distinct biological pathways/processes (e.g., cell cycle for STA and immune diseases and calcium signalling pathway for LTA). Epigenetic clock analysis revealed evidence of accelerated ageing in the acclimatized newcomers compared to the native lowlanders. Our research provides novel insights into epigenetic regulation in relation to high altitude and intervention strategies for altitude-related ageing or illnesses.
© 2025. The Author(s).
Conflict of interest statement
Conflict of interest: The authors declare no competing interests.
Figures





Similar articles
-
A narrative review of periodic breathing during sleep at high altitude: From acclimatizing lowlanders to adapted highlanders.J Physiol. 2024 Nov;602(21):5435-5448. doi: 10.1113/JP285427. Epub 2024 Mar 27. J Physiol. 2024. PMID: 38534039 Review.
-
Echocardiographic and tissue Doppler imaging of cardiac adaptation to high altitude in native highlanders versus acclimatized lowlanders.Am J Cardiol. 2009 Jun 1;103(11):1605-9. doi: 10.1016/j.amjcard.2009.02.006. Epub 2009 Apr 8. Am J Cardiol. 2009. PMID: 19463523
-
Comparison of Echocardiographic Parameters Between Healthy Highlanders in Tibet and Lowlanders in Beijing.High Alt Med Biol. 2018 Sep;19(3):259-264. doi: 10.1089/ham.2017.0094. Epub 2018 Aug 17. High Alt Med Biol. 2018. PMID: 30118327
-
Left ventricular adaptation to high altitude: speckle tracking echocardiography in lowlanders, healthy highlanders and highlanders with chronic mountain sickness.Int J Cardiovasc Imaging. 2015 Apr;31(4):743-52. doi: 10.1007/s10554-015-0614-1. Epub 2015 Feb 10. Int J Cardiovasc Imaging. 2015. PMID: 25665685
-
The impact of hypoxaemia on vascular function in lowlanders and high altitude indigenous populations.J Physiol. 2019 Dec;597(24):5759-5776. doi: 10.1113/JP277191. Epub 2019 Nov 28. J Physiol. 2019. PMID: 31677355 Review.
Cited by
-
DNA methylation in adaptation to high-altitude environments and pathogenesis of related diseases.Hum Genomics. 2025 Aug 30;19(1):100. doi: 10.1186/s40246-025-00794-x. Hum Genomics. 2025. PMID: 40886024 Free PMC article. Review.
References
-
- Körner, C. The use of ‘altitude’ in ecological research. Trends Ecol. Evol.22, 569–574 (2007). - PubMed
-
- Zhang, X. L. et al. The earliest human occupation of the high-altitude Tibetan Plateau 40 thousand to 30 thousand years ago. Science362, 1049–1051 (2018). - PubMed
-
- Bennett, A., Sain, S. R., Vargas, E. & Moore, L. G. Evidence that parent-of-origin affects birth-weight reductions at high altitude. Am. J. Hum. Biol.20, 592–597 (2008). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
