

Longitudinal profiling of low-abundance strains in microbiomes with ChronoStrain
- PMID: 40328944
- PMCID: PMC12122369
- DOI: 10.1038/s41564-025-01983-z
Longitudinal profiling of low-abundance strains in microbiomes with ChronoStrain
Erratum in
-
Author Correction: Longitudinal profiling of low-abundance strains in microbiomes with ChronoStrain.Nat Microbiol. 2025 Jun;10(6):1551. doi: 10.1038/s41564-025-02042-3. Nat Microbiol. 2025. PMID: 40442503 Free PMC article. No abstract available.
Abstract
The ability to detect and quantify microbiota over time from shotgun metagenomic data has a plethora of clinical, basic science and public health applications. Given these applications, and the observation that pathogens and other taxa of interest can reside at low relative abundance, there is a critical need for algorithms that accurately profile low-abundance microbial taxa with strain-level resolution. Here we present ChronoStrain: a sequence quality- and time-aware Bayesian model for profiling strains in longitudinal samples. ChronoStrain explicitly models the presence or absence of each strain and produces a probability distribution over abundance trajectories for each strain. Using synthetic and semi-synthetic data, we demonstrate how ChronoStrain outperforms existing methods in abundance estimation and presence/absence prediction. Applying ChronoStrain to two human microbiome datasets demonstrated its improved interpretability for profiling Escherichia coli strain blooms in longitudinal faecal samples from adult women with recurring urinary tract infections, and its improved accuracy for detecting Enterococcus faecalis strains in infant faecal samples. Compared with state-of-the-art methods, ChronoStrain's ability to detect low-abundance taxa is particularly stark.
© 2025. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures

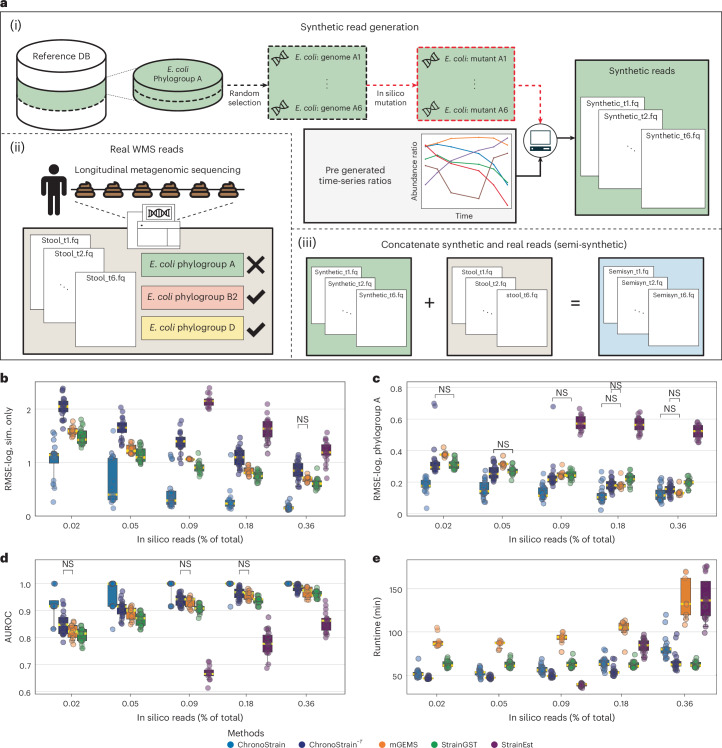
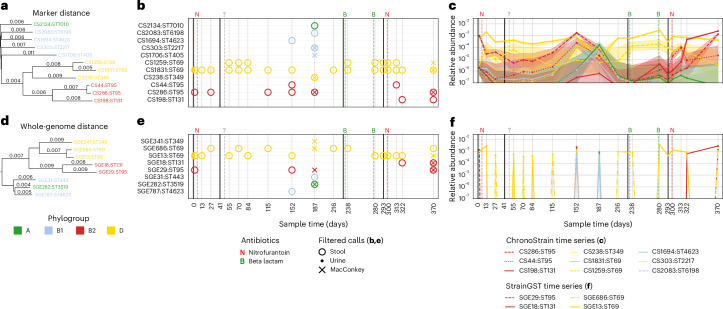










Update of
-
Strain tracking with uncertainty quantification.bioRxiv [Preprint]. 2024 Jul 23:2023.01.25.525531. doi: 10.1101/2023.01.25.525531. bioRxiv. 2024. Update in: Nat Microbiol. 2025 May;10(5):1184-1197. doi: 10.1038/s41564-025-01983-z. PMID: 36747646 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
- R35 GM149270/GM/NIGMS NIH HHS/United States
- R35GM141861/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 GM130777/GM/NIGMS NIH HHS/United States
- R01DK121822/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R21 AI154075/AI/NIAID NIH HHS/United States
- R35 GM141861/GM/NIGMS NIH HHS/United States
- U19 AI110818/AI/NIAID NIH HHS/United States
- R21AI154075/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- U19AI110818/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R35 GM143056/GM/NIGMS NIH HHS/United States
- R35GM149270/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 DK121822/DK/NIDDK NIH HHS/United States
- R35GM143056/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
