

Simultaneous detection of pathogens and antimicrobial resistance genes with the open source, cloud-based, CZ ID platform
- PMID: 40329334
- PMCID: PMC12057172
- DOI: 10.1186/s13073-025-01480-2
Simultaneous detection of pathogens and antimicrobial resistance genes with the open source, cloud-based, CZ ID platform
Abstract
Background: Antimicrobial resistant (AMR) pathogens represent urgent threats to human health, and their surveillance is of paramount importance. Metagenomic next-generation sequencing (mNGS) has revolutionized such efforts, but remains challenging due to the lack of open-access bioinformatics tools capable of simultaneously analyzing both microbial and AMR gene sequences.
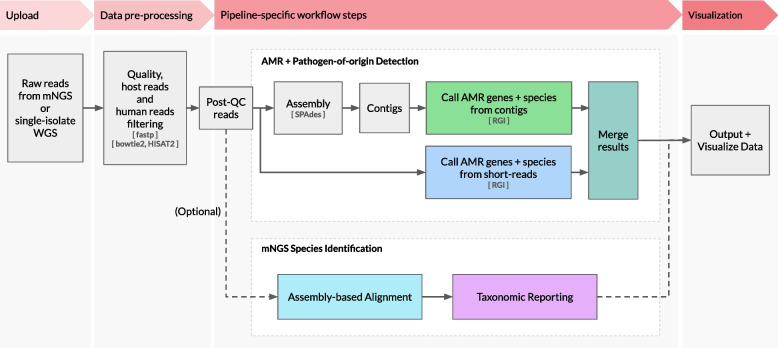
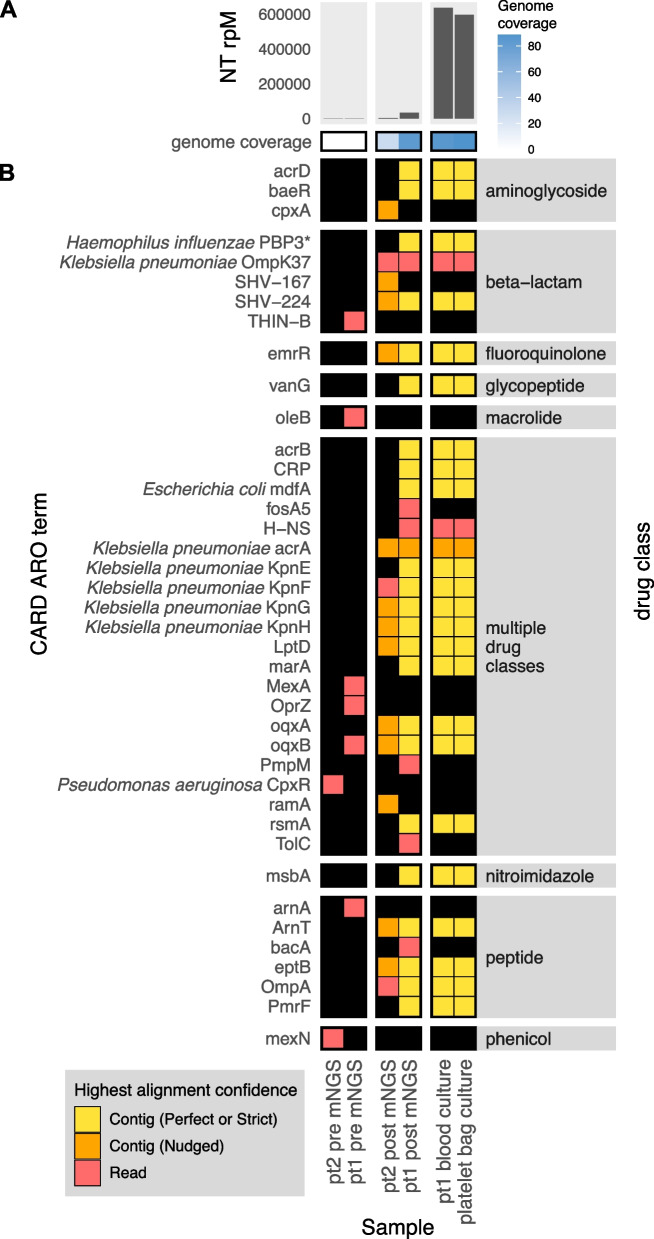
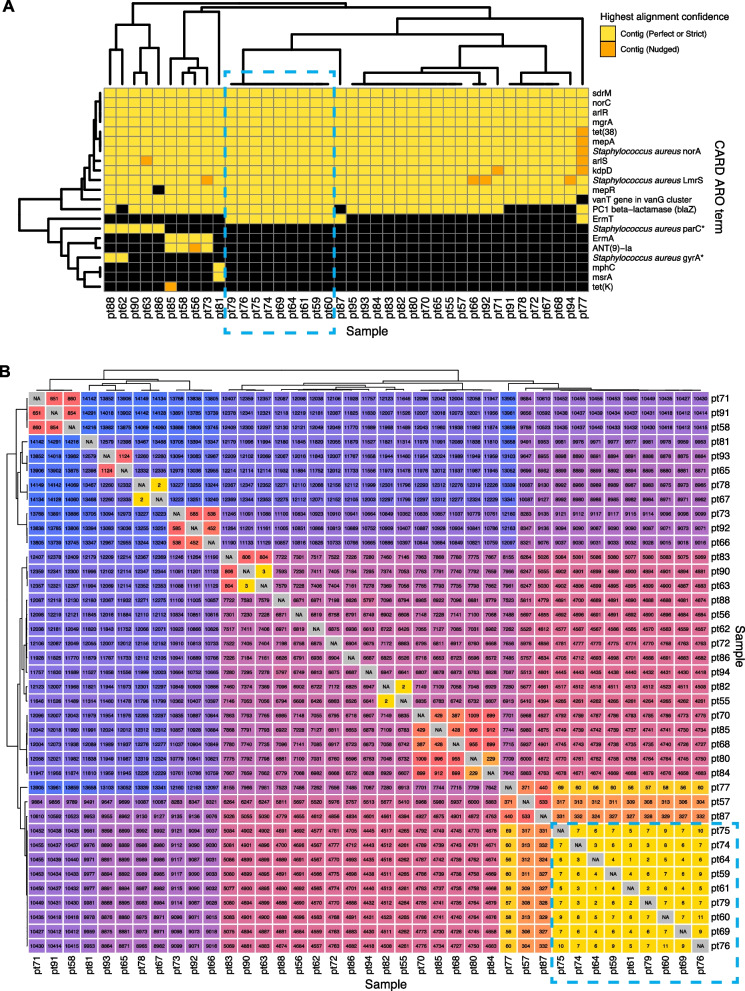
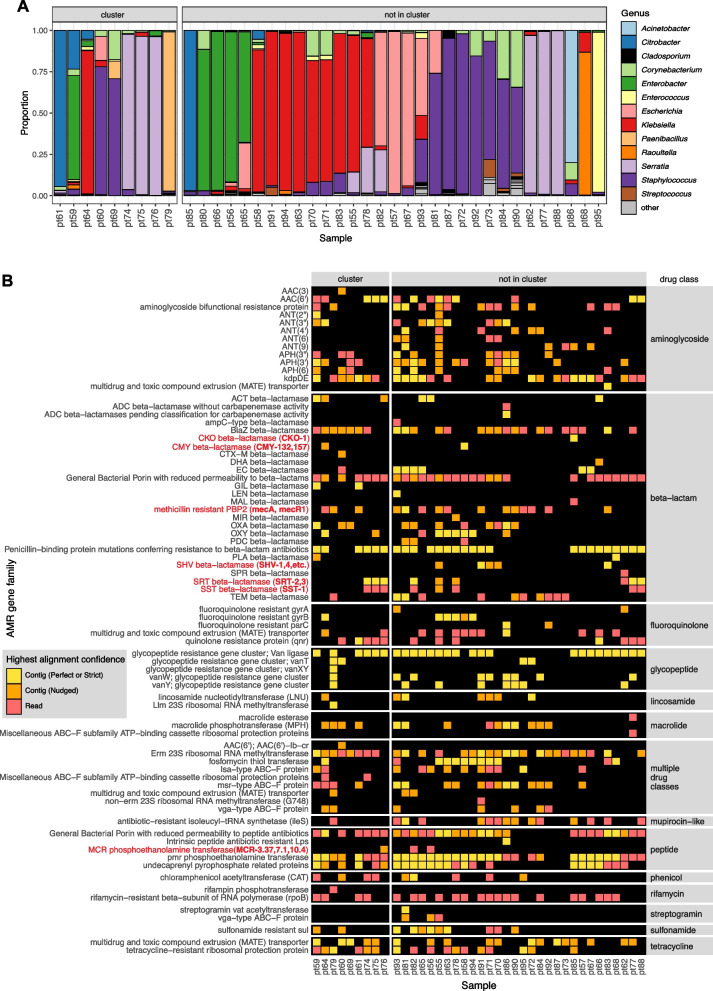
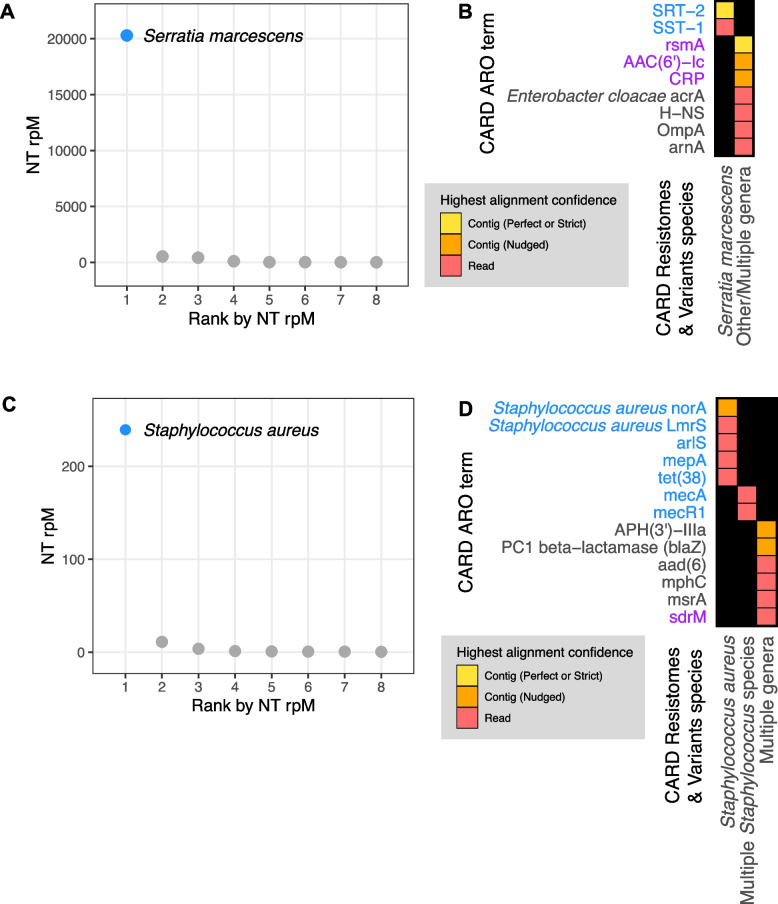
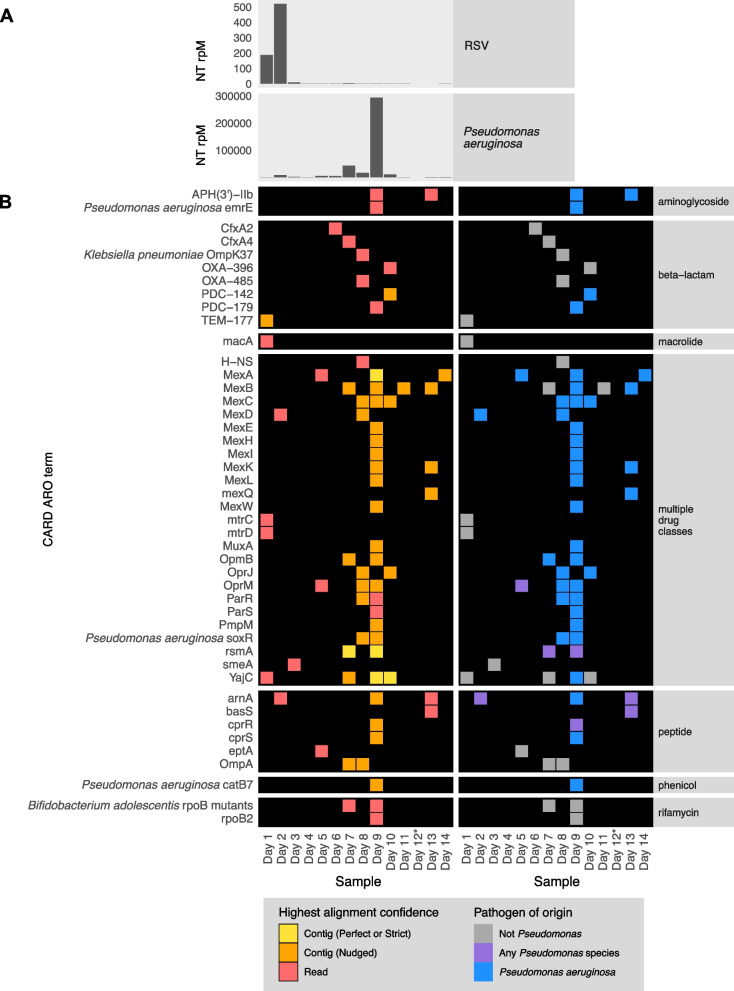
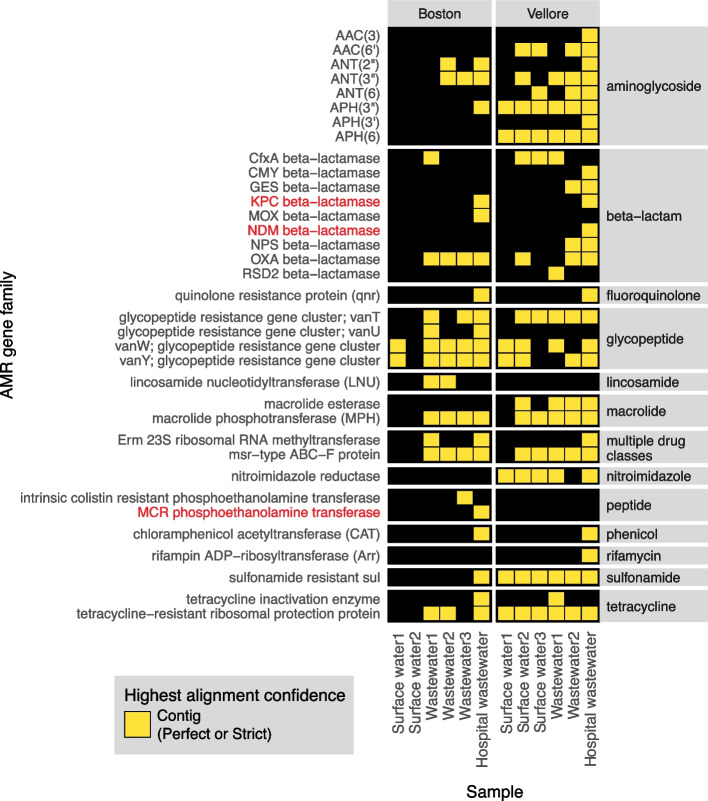
Results: To address this need, we developed the Chan Zuckerberg ID (CZ ID) AMR module, an open-access, cloud-based workflow designed to integrate detection of both microbes and AMR genes in mNGS and single-isolate whole-genome sequencing (WGS) data. It leverages the Comprehensive Antibiotic Resistance Database and associated Resistance Gene Identifier software, and works synergistically with the CZ ID short-read mNGS module to enable broad detection of both microbes and AMR genes from Illumina data. We highlight diverse applications of the AMR module through analysis of both publicly available and newly generated mNGS and single-isolate WGS data from four clinical cohort studies and an environmental surveillance project. Through genomic investigations of bacterial sepsis and pneumonia cases, hospital outbreaks, and wastewater surveillance data, we gain a deeper understanding of infectious agents and their resistomes, highlighting the value of integrating microbial identification and AMR profiling for both research and public health. We leverage additional functionalities of the CZ ID mNGS platform to couple resistome profiling with the assessment of phylogenetic relationships between nosocomial pathogens, and further demonstrate the potential to capture the longitudinal dynamics of pathogen and AMR genes in hospital acquired bacterial infections.
Conclusions: In sum, the new AMR module advances the capabilities of the open-access CZ ID microbial bioinformatics platform by integrating pathogen detection and AMR profiling from mNGS and single-isolate WGS data. Its development represents an important step toward democratizing pathogen genomic analysis and supporting collaborative efforts to combat the growing threat of AMR.
Keywords: Antimicrobial resistance; CZ ID; Chan Zuckerberg ID; Metagenomics; Whole-genome sequencing.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This research conformed to the principles of the Helsinki Declaration. Skin swabs and cultured isolates analyzed for Application 2 (hospital outbreak) were collected under the University of California San Francisco (UCSF) Institutional Review Board (IRB) protocol no. 17–24056. A waiver of consent was granted for swab collection given that the primary purpose was for monitoring and preventing transmission of healthcare-associated pathogens in the hospital, in coordination with the UCSF Department of Hospital Epidemiology and Infection Prevention. Samples analyzed for Application 4 (longitudinal profiling) were collected from patients enrolled in a prospective cohort study of mechanically ventilated children admitted to eight intensive care units in the National Institute of Child Health and Human Development’s Collaborative Pediatric Critical Care Research Network (CPCCRN) from February 2015 to December 2017. The original cohort study was approved by the Collaborative Pediatric Critical Care Research IRB at the University of Utah (protocol no. 00088656). Details regarding enrollment and consent have previously been described [38, 39]. Briefly, children aged 31 days to 18 years who were expected to require mechanical ventilation via endotracheal tube for at least 72 h were enrolled. Parents or other legal guardians of eligible patients were approached for consent by study-trained staff as soon as possible after intubation. To permit sample collection as early as possible following intensive care unit admission, the IRB granted an initial waiver of consent for the collection of endotracheal aspirate by standard-of-care suctioning. Parents or guardians were then approached for informed consent, and samples were only retained and analyzed if written informed consent for participation in the study was provided. For all other applications and analyses, previously published datasets were used as described in the data and code availability section. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures

Update of
-
Simultaneous detection of pathogens and antimicrobial resistance genes with the open source, cloud-based, CZ ID pipeline.bioRxiv [Preprint]. 2024 Apr 18:2024.04.12.589250. doi: 10.1101/2024.04.12.589250. bioRxiv. 2024. Update in: Genome Med. 2025 May 6;17(1):46. doi: 10.1186/s13073-025-01480-2. PMID: 38645206 Free PMC article. Updated. Preprint.
-
Simultaneous detection of pathogens and antimicrobial resistance genes with the open source, cloud-based, CZ ID pipeline.Res Sq [Preprint]. 2024 May 2:rs.3.rs-4271356. doi: 10.21203/rs.3.rs-4271356/v1. Res Sq. 2024. Update in: Genome Med. 2025 May 6;17(1):46. doi: 10.1186/s13073-025-01480-2. PMID: 38746293 Free PMC article. Updated. Preprint.
References
-
- O’Neill J. Tackling drug-resistant infections globally: final report and recommendations. Government of the United Kingdom. 2016. Available from: https://amr-review.org/Publications.html.
-
- 10 global health issues to track in 2021. Available from: https://www.who.int/news-room/spotlight/10-global-health-issues-to-track.... Cited 2024 May 31.
-
- Baker KS, Jauneikaite E, Nunn JG, Midega JT, Atun R, Holt KE, et al. Evidence review and recommendations for the implementation of genomics for antimicrobial resistance surveillance: reports from an international expert group. Lancet Microbe. 2023;4:e1035–9. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
