

The CpG Landscape of Protein Coding DNA in Vertebrates
- PMID: 40330995
- PMCID: PMC12050414
- DOI: 10.1111/eva.70101
The CpG Landscape of Protein Coding DNA in Vertebrates
Abstract
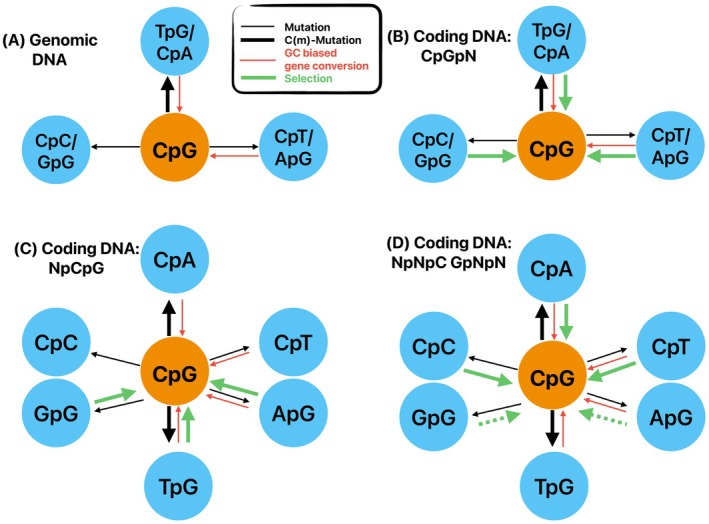
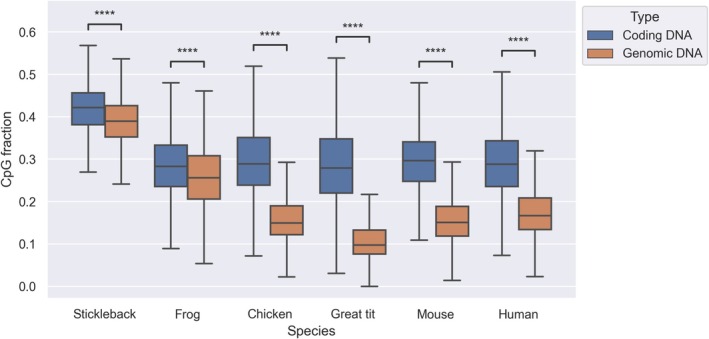
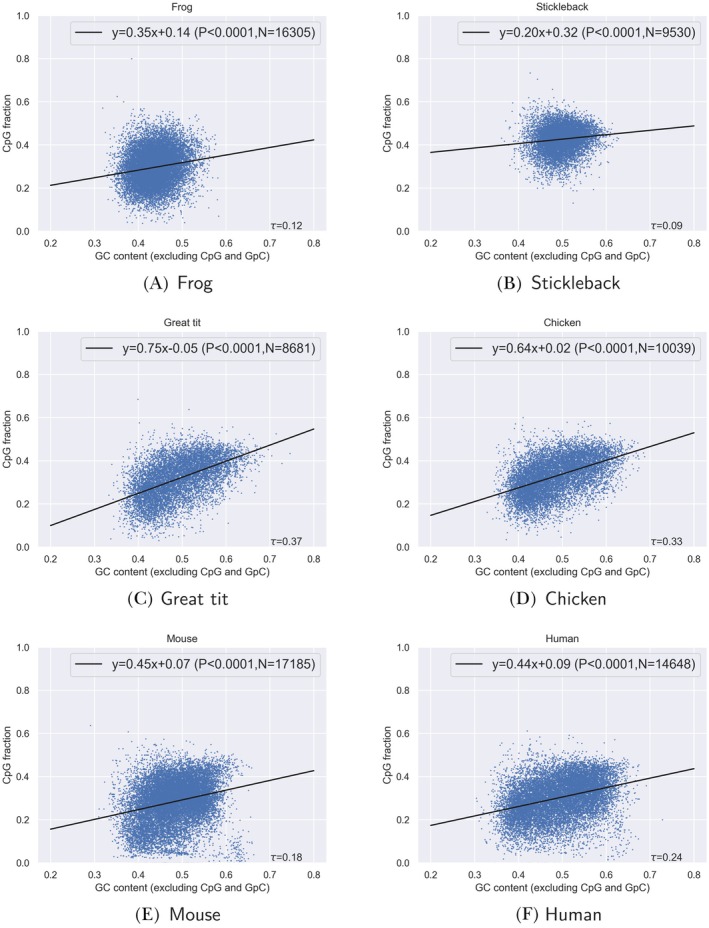
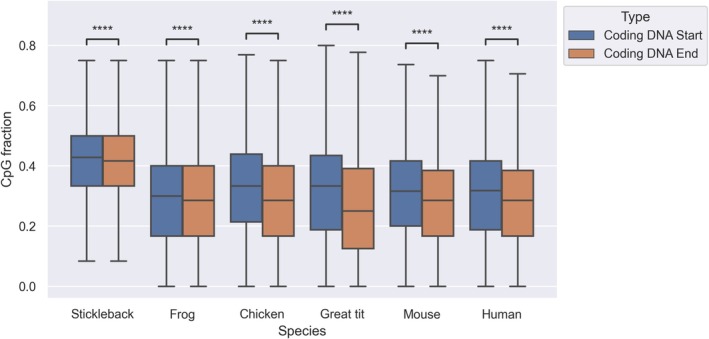
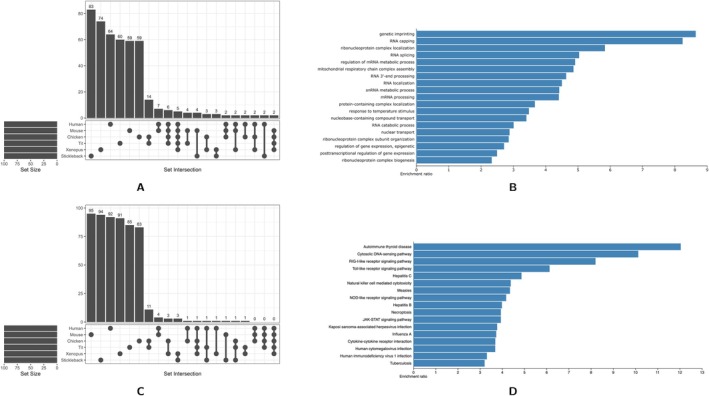
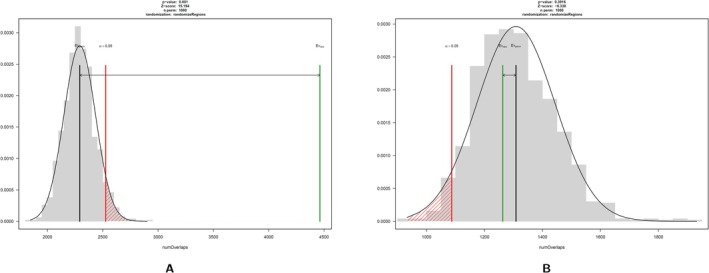
DNA methylation has fundamental implications for vertebrate genome evolution by influencing the mutational landscape, particularly at CpG dinucleotides. Methylation-induced mutations drive a genome-wide depletion of CpG sites, creating a dinucleotide composition bias across the genome. Examination of the standard genetic code reveals CpG to be the only facultative dinucleotide; it is however unclear what specific implications CpG bias has on protein coding DNA. Here, we use theoretical considerations of the genetic code combined with empirical genome-wide analyses in six vertebrate species-human, mouse, chicken, great tit, frog, and stickleback-to investigate how CpG content is shaped and maintained in protein-coding genes. We show that protein-coding sequences consistently exhibit significantly higher CpG content than noncoding regions and demonstrate that CpG sites are enriched in genes involved in regulatory functions and stress responses, suggesting selective maintenance of CpG content in specific loci. These findings have important implications for evolutionary applications in both natural and managed populations: CpG content could serve as a genetic marker for assessing adaptive potential, while the identification of CpG-free codons provides a framework for genome optimization in breeding and synthetic biology. Our results underscore the intricate interplay between mutational biases, selection, and epigenetic regulation, offering new insights into how vertebrate genomes evolve under varying ecological and selective pressures.
Keywords: DNA methylation; base composition; dinucleotides; epigenetics; protein coding DNA.
© 2025 The Author(s). Evolutionary Applications published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures


Similar articles
-
Modelling mutational and selection pressures on dinucleotides in eukaryotic phyla--selection against CpG and UpA in cytoplasmically expressed RNA and in RNA viruses.BMC Genomics. 2013 Sep 10;14:610. doi: 10.1186/1471-2164-14-610. BMC Genomics. 2013. PMID: 24020411 Free PMC article.
-
Depletion of CpG Dinucleotides in Papillomaviruses and Polyomaviruses: A Role for Divergent Evolutionary Pressures.PLoS One. 2015 Nov 6;10(11):e0142368. doi: 10.1371/journal.pone.0142368. eCollection 2015. PLoS One. 2015. PMID: 26544572 Free PMC article.
-
CpG islands in vertebrate genomes.J Mol Biol. 1987 Jul 20;196(2):261-82. doi: 10.1016/0022-2836(87)90689-9. J Mol Biol. 1987. PMID: 3656447
-
Sequence determinants, function, and evolution of CpG islands.Biochem Soc Trans. 2021 Jun 30;49(3):1109-1119. doi: 10.1042/BST20200695. Biochem Soc Trans. 2021. PMID: 34156435 Free PMC article. Review.
-
CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function.Genes (Basel). 2017 May 23;8(6):148. doi: 10.3390/genes8060148. Genes (Basel). 2017. PMID: 28545252 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
