

Establishment of a Human iPSC Line from Mucolipidosis Type II That Expresses the Key Markers of the Disease
- PMID: 40332602
- PMCID: PMC12027929
- DOI: 10.3390/ijms26083871
Establishment of a Human iPSC Line from Mucolipidosis Type II That Expresses the Key Markers of the Disease
Abstract
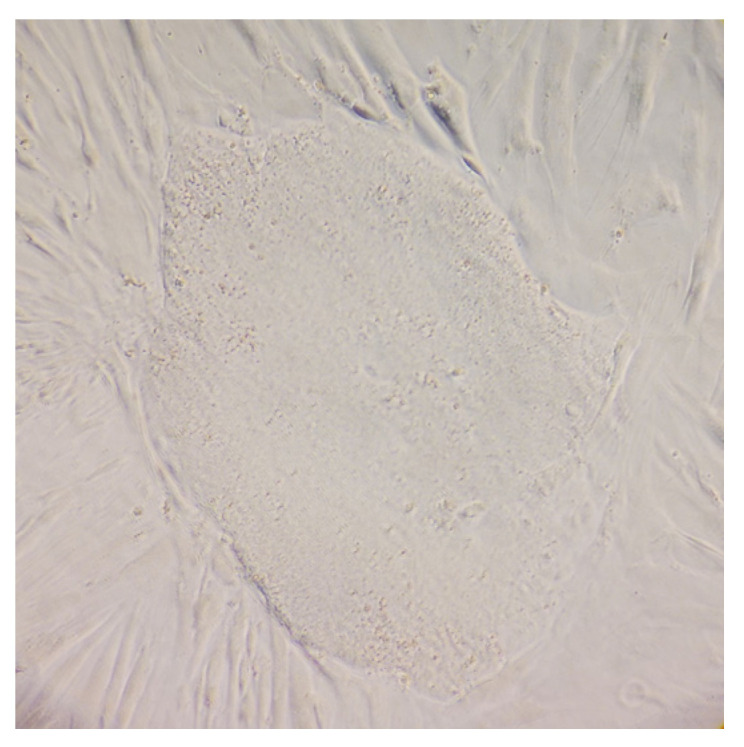
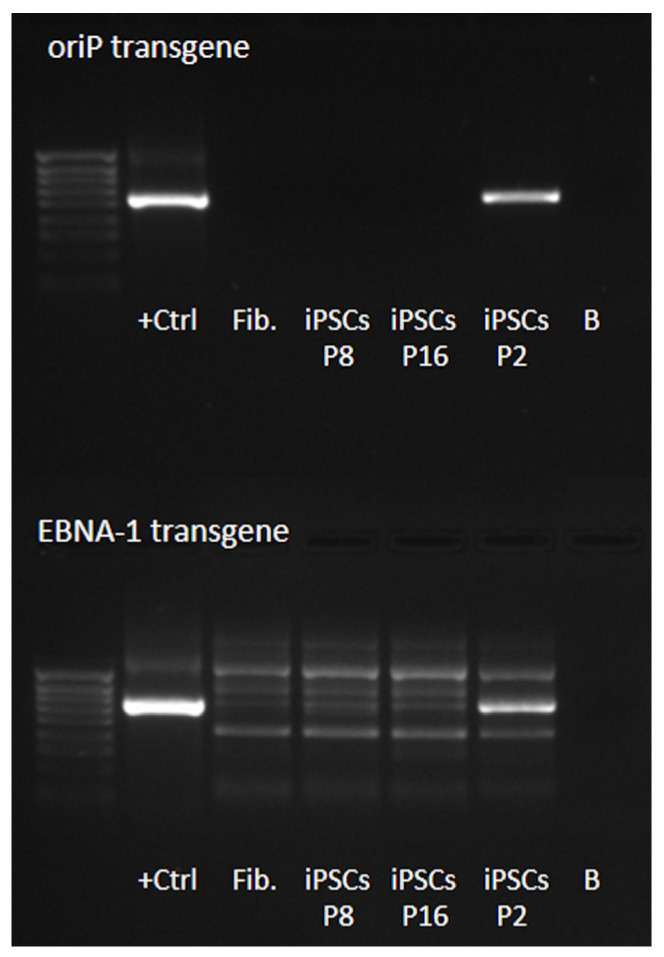
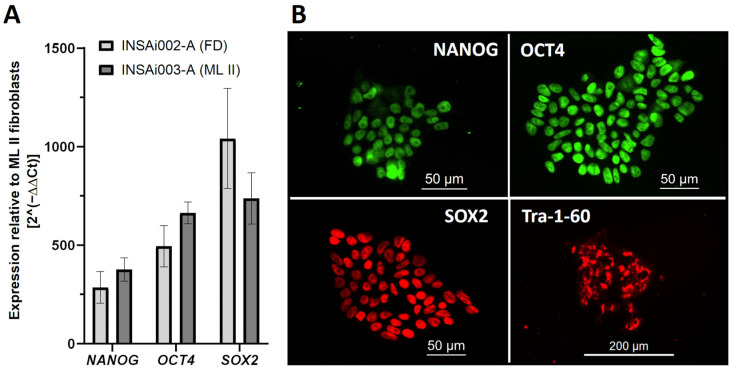
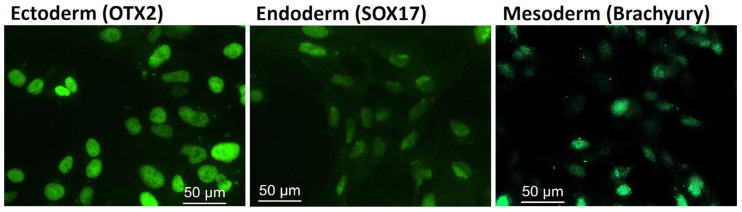
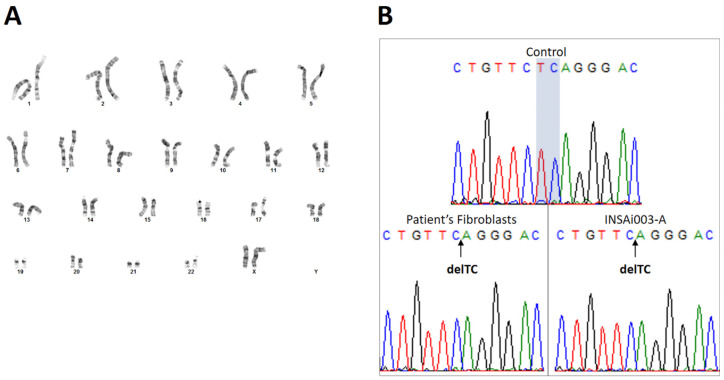
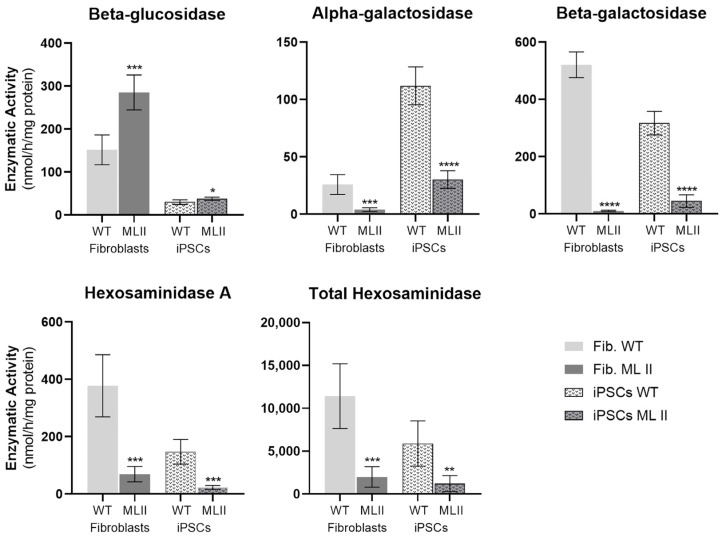
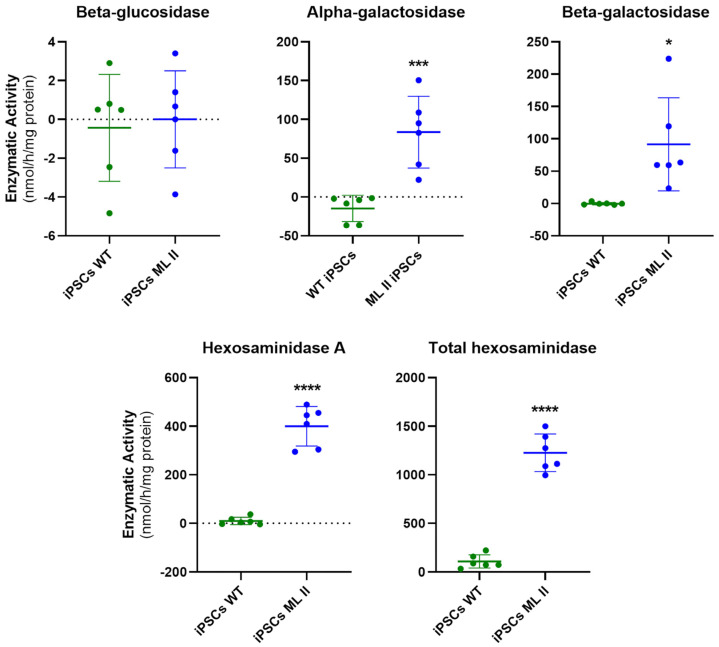
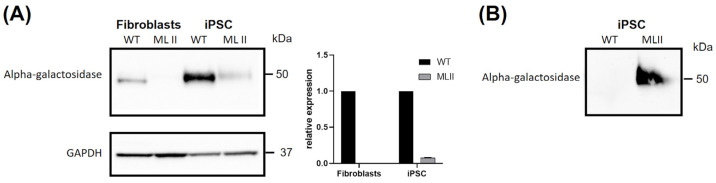
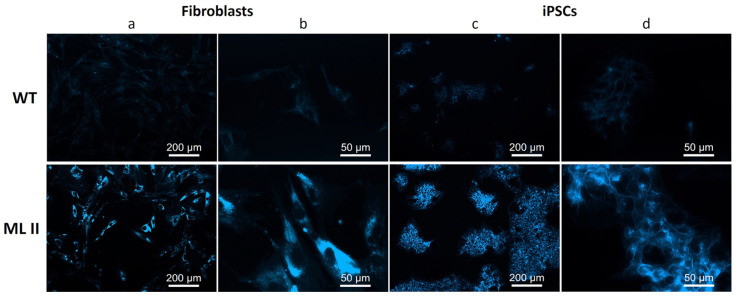
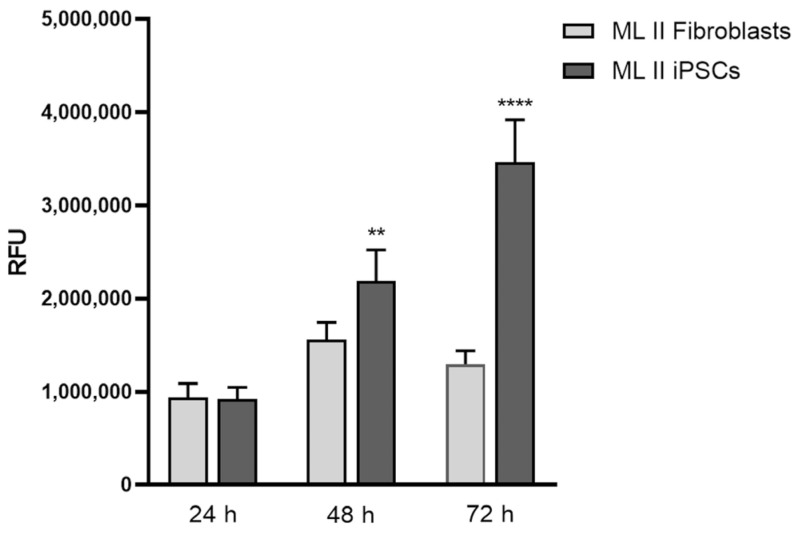
Mucolipidosis type II (ML II) is a rare and fatal disease of acid hydrolase trafficking. It is caused by pathogenic variants in the GNPTAB gene, leading to the absence of GlcNAc-1-phosphotransferase activity, an enzyme that catalyzes the first step in the formation of the mannose 6-phosphate (M6P) tag, essential for the trafficking of most lysosomal hydrolases. Without M6P, these do not reach the lysosome, which accumulates undegraded substrates. The lack of samples and adequate disease models limits the investigation into the pathophysiological mechanisms of the disease and potential therapies. Here, we report the generation and characterization of an ML II induced pluripotent stem cell (iPSC) line carrying the most frequent ML II pathogenic variant [NM_024312.5(GNPTAB):c.3503_3504del (p.Leu1168fs)]. Skin fibroblasts were successfully reprogrammed into iPSCs that express pluripotency markers, maintain a normal karyotype, and can differentiate into the three germ layers. Furthermore, ML II iPSCs showed a phenotype comparable to that of the somatic cells that originated them in terms of key ML II hallmarks: lower enzymatic activity of M6P-dependent hydrolases inside the cells but higher in conditioned media, and no differences in an M6P-independent hydrolase and accumulation of free cholesterol. Thus, ML II iPSCs constitute a novel model for ML II disease, with the inherent iPSC potential to become a valuable model for future studies on the pathogenic mechanisms and testing potential therapeutic approaches.
Keywords: LSD; ML II; cellular model; iPSCs; lysosomal storage disorder; stem cells.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Dogterom E.J., Wagenmakers M.A.E.M., Wilke M., Demirdas S., Muschol N.M., Pohl S., Meijden J.C.v.d., Rizopoulos D., Ploeg A.T.v.d., Oussoren E. Mucolipidosis Type II and Type III: A Systematic Review of 843 Published Cases. Genet. Med. 2021;23:2047–2056. doi: 10.1038/s41436-021-01244-4. - DOI - PubMed
-
- Velho R.V., Harms F.L., Danyukova T., Ludwig N.F., Friez M.J., Cathey S.S., Filocamo M., Tappino B., Güneş N., Tüysüz B., et al. The Lysosomal Storage Disorders Mucolipidosis Type II, Type III Alpha/Beta, and Type III Gamma: Update on GNPTAB and GNPTG Mutations. Hum. Mutat. 2019;40:842–864. doi: 10.1002/HUMU.23748. - DOI - PubMed
-
- Matos L., Vilela R., Rocha M., Santos J.I., Coutinho M.F., Gaspar P., Prata M.J., Alves S. Development of an Antisense Oligonucleotide-Mediated Exon Skipping Therapeutic Strategy for Mucolipidosis II: Validation at RNA Level. Hum. Gene Ther. 2020;31:775–783. doi: 10.1089/hum.2020.034. - DOI - PubMed
-
- Coutinho M., Encarnação M., Gomes R., da Silva Santos L., Martins S., Sirois-Gagnon D., Bargal R., Filocamo M., Raas-Rothschild A., Tappino B., et al. Origin and Spread of a Common Deletion Causing Mucolipidosis Type II: Insights from Patterns of Haplotypic Diversity. Clin. Genet. 2011;80:273–280. doi: 10.1111/j.1399-0004.2010.01539.x. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
