

Native nucleosomes intrinsically encode genome organization principles
- PMID: 40335690
- PMCID: PMC12240700
- DOI: 10.1038/s41586-025-08971-7
Native nucleosomes intrinsically encode genome organization principles
Abstract
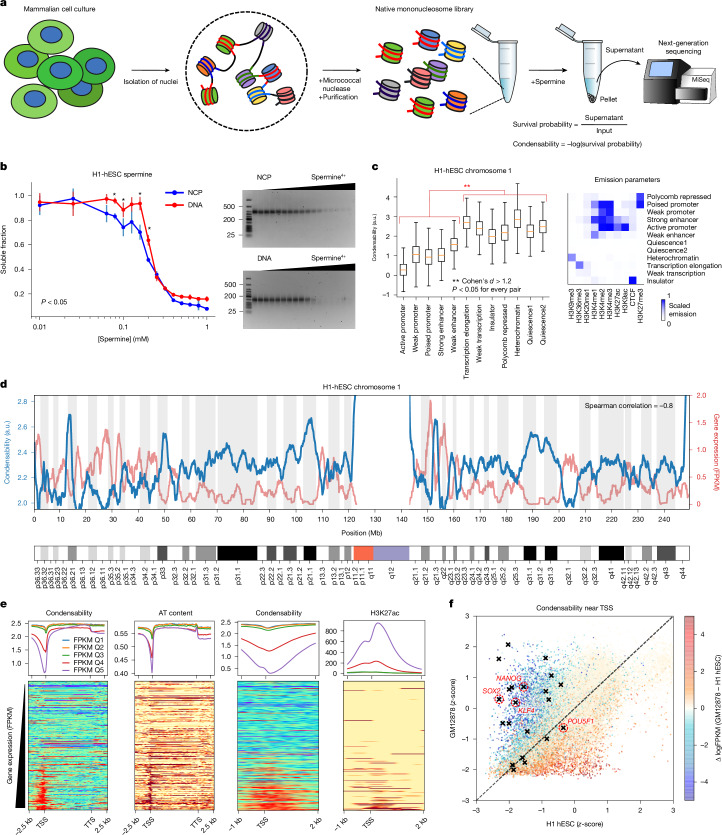
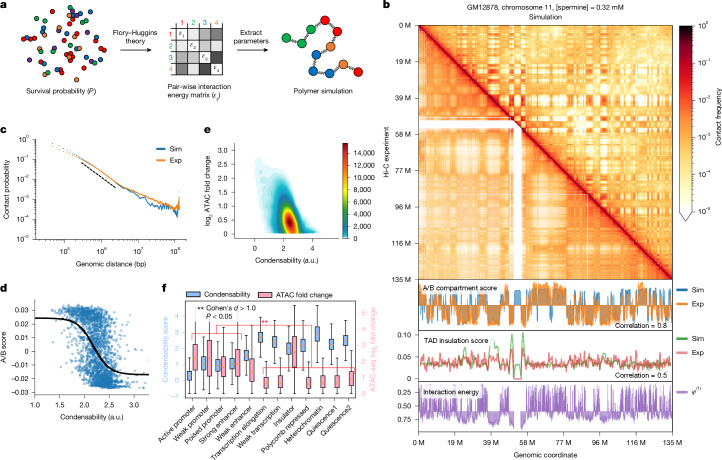
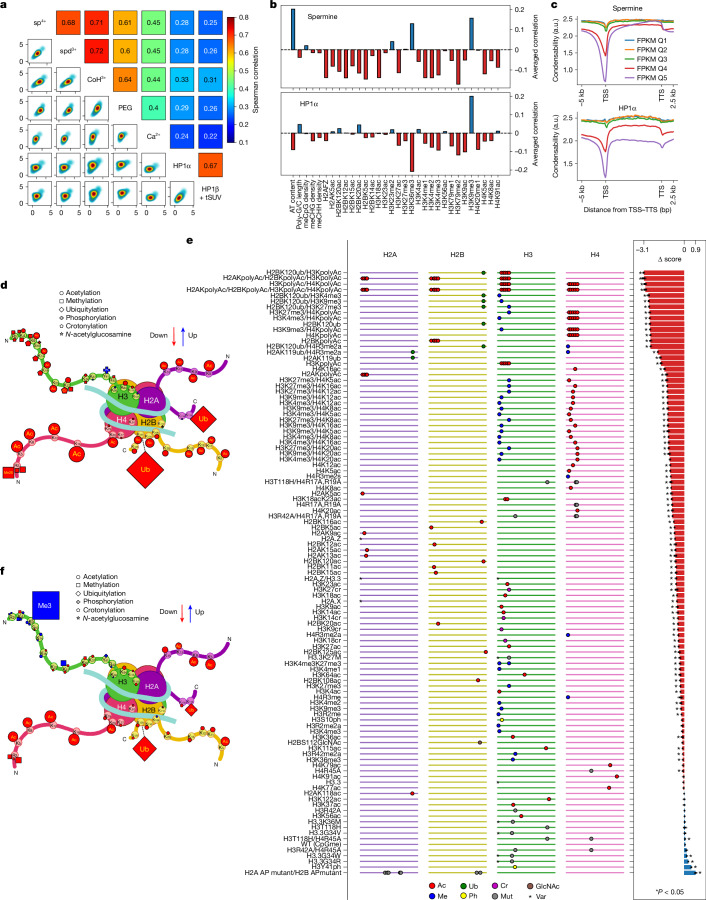
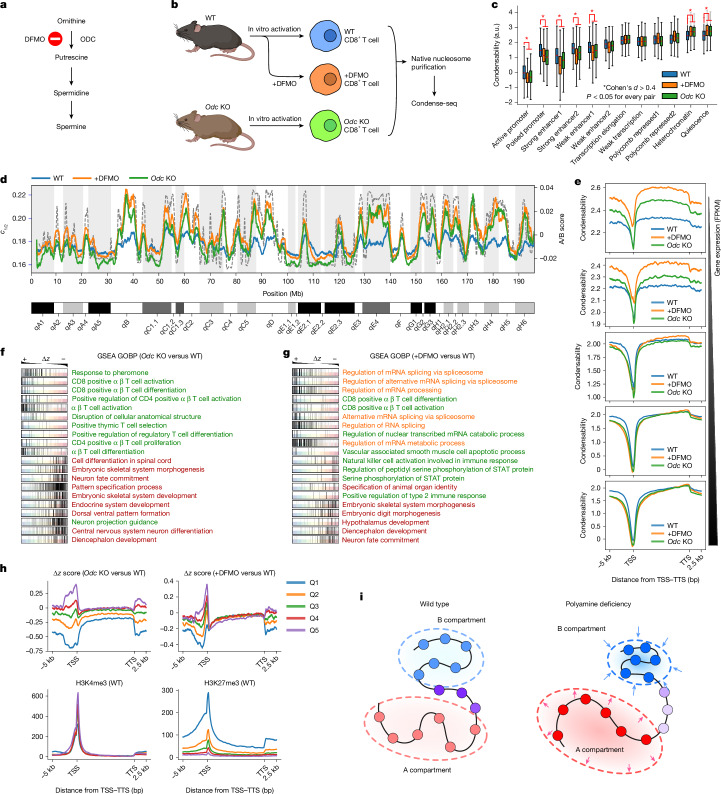
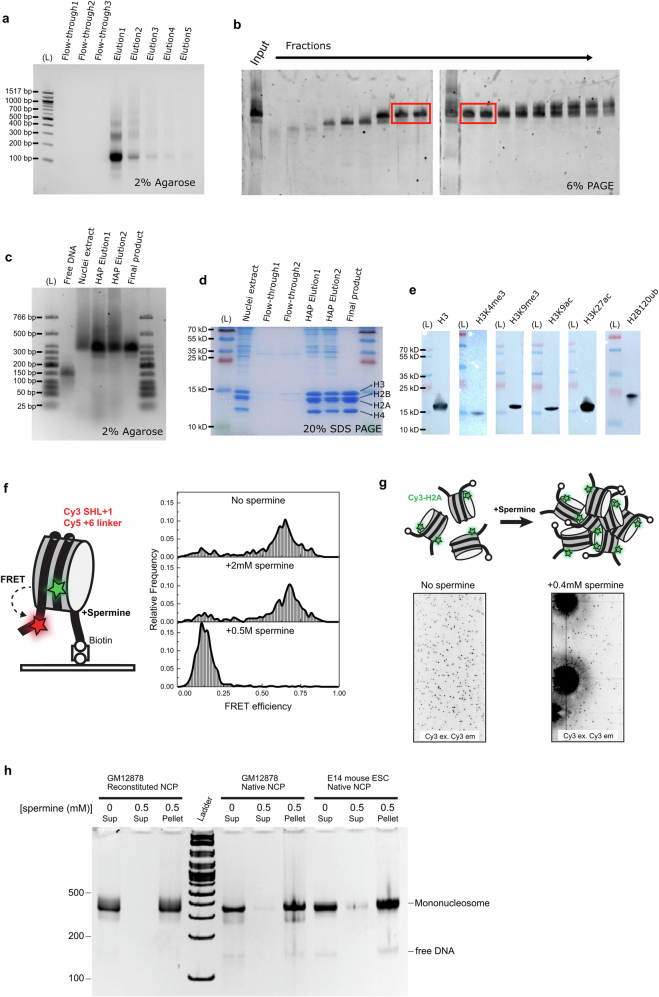
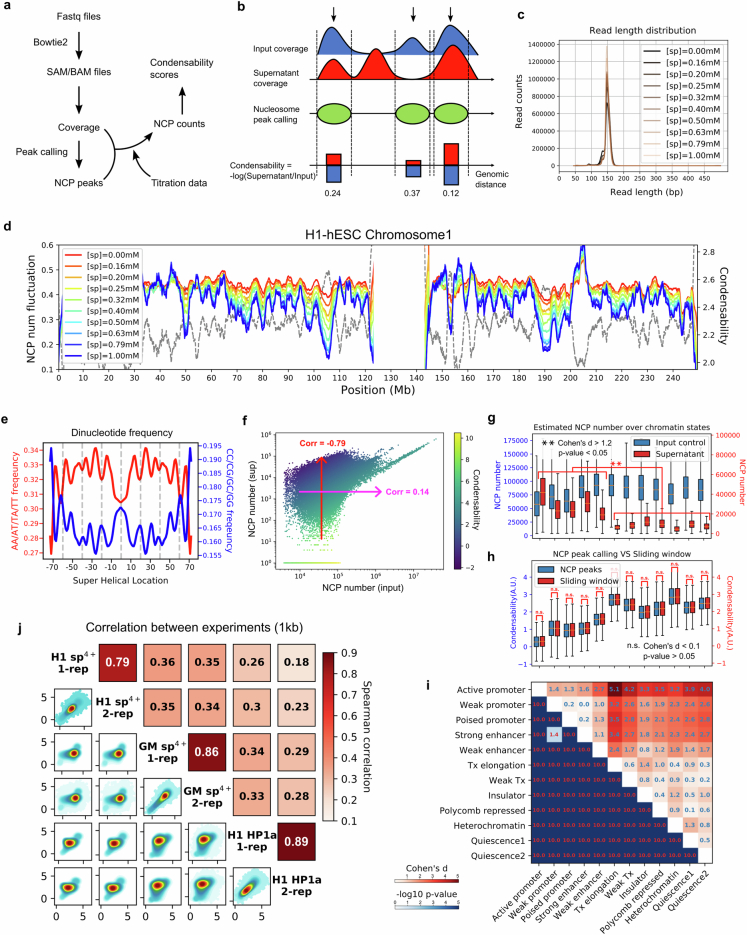
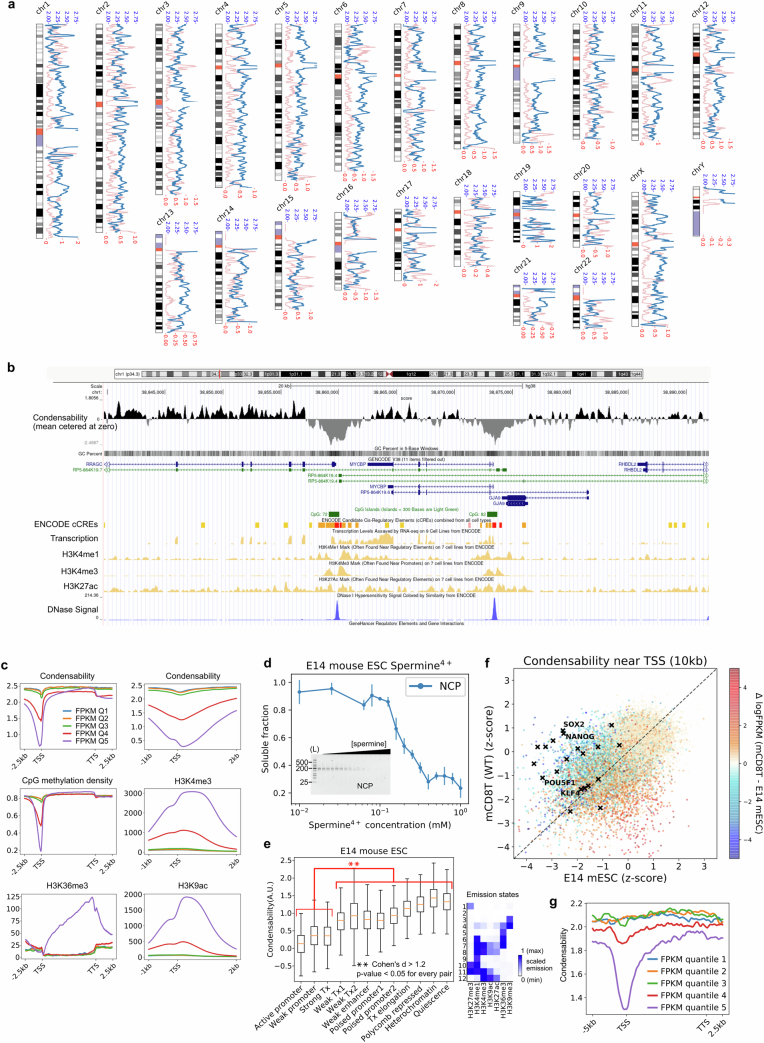
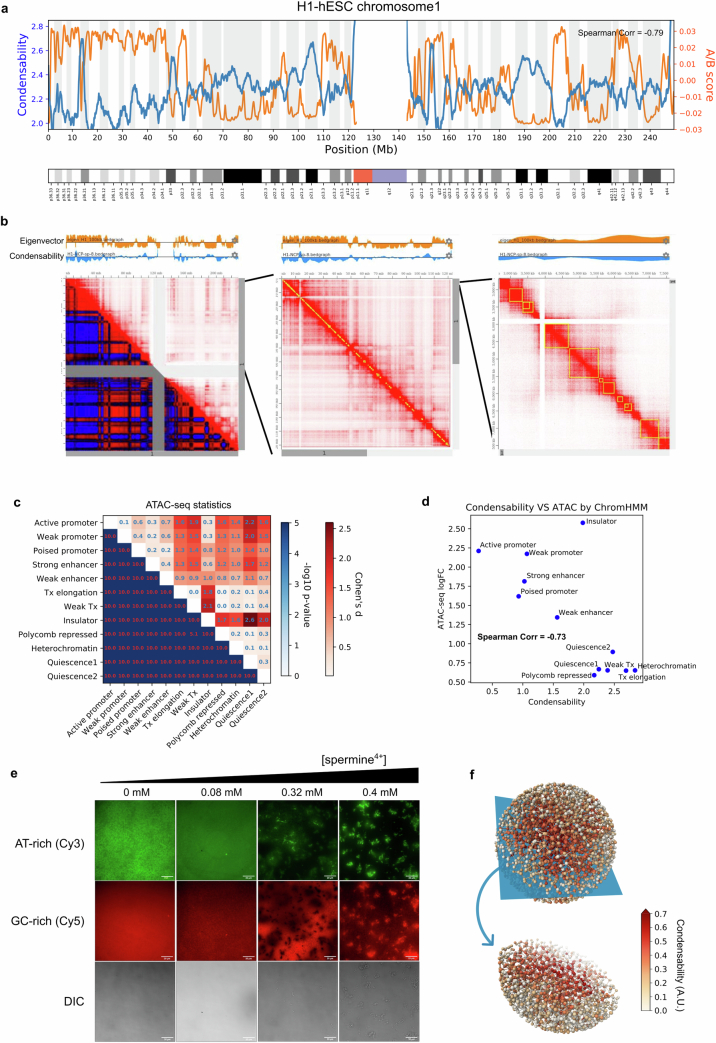
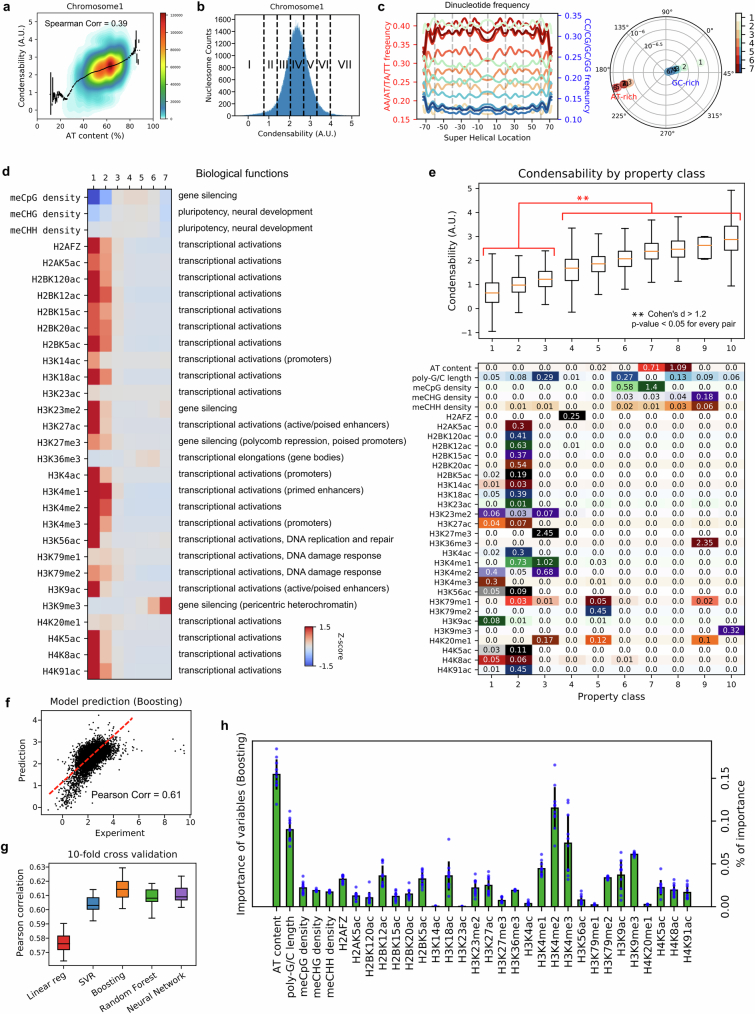
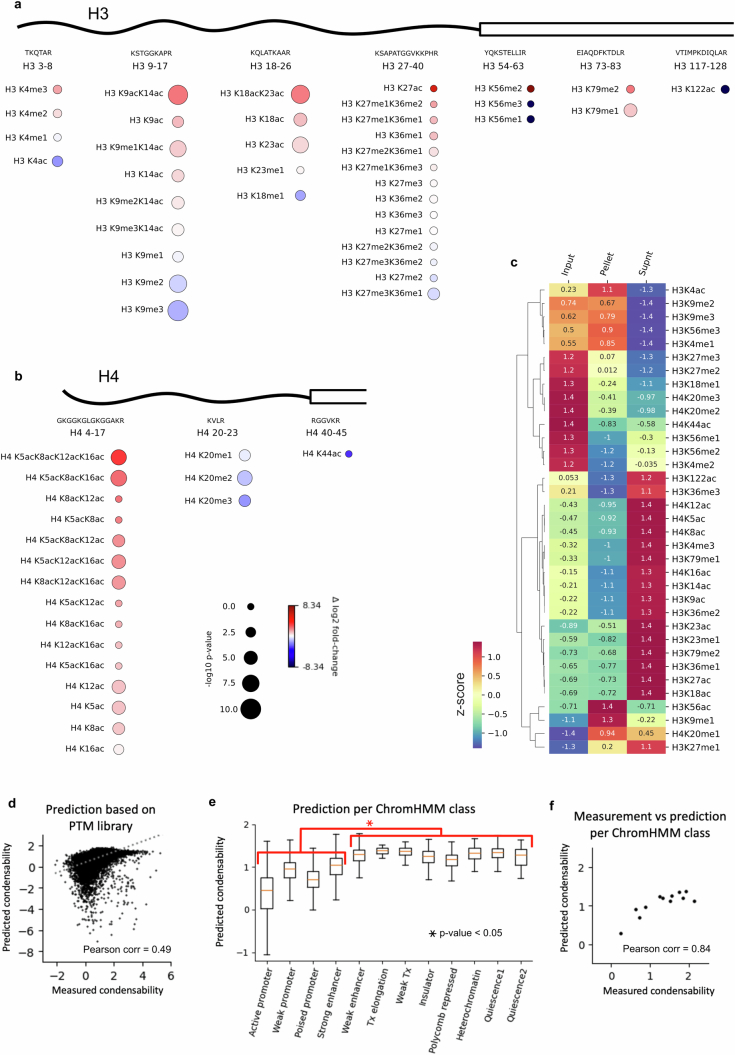
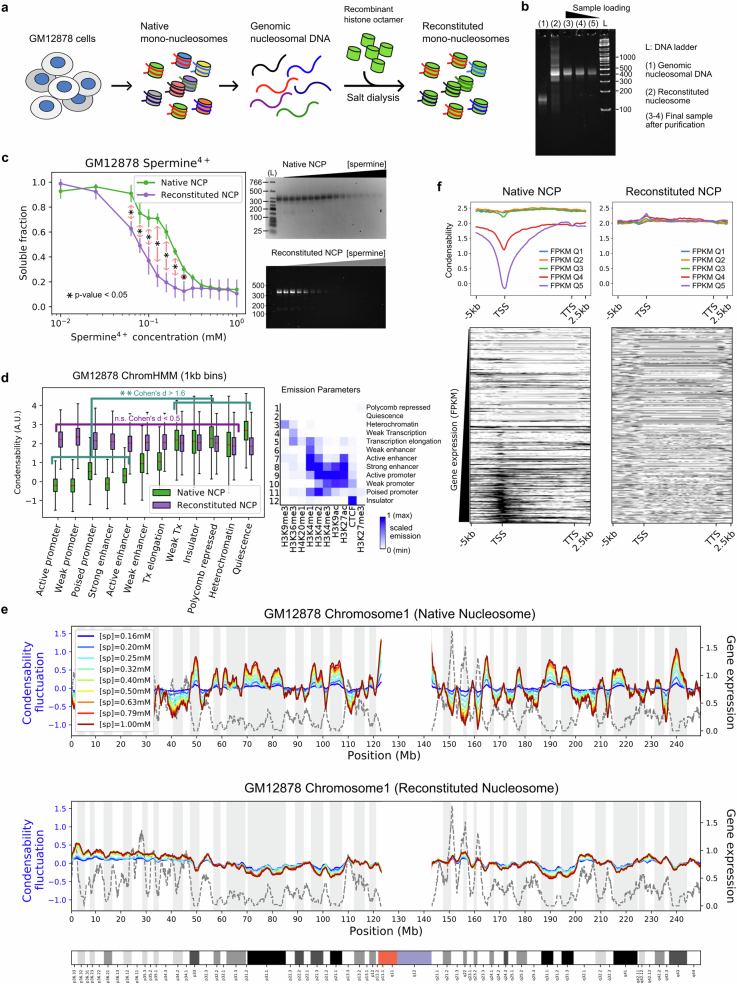
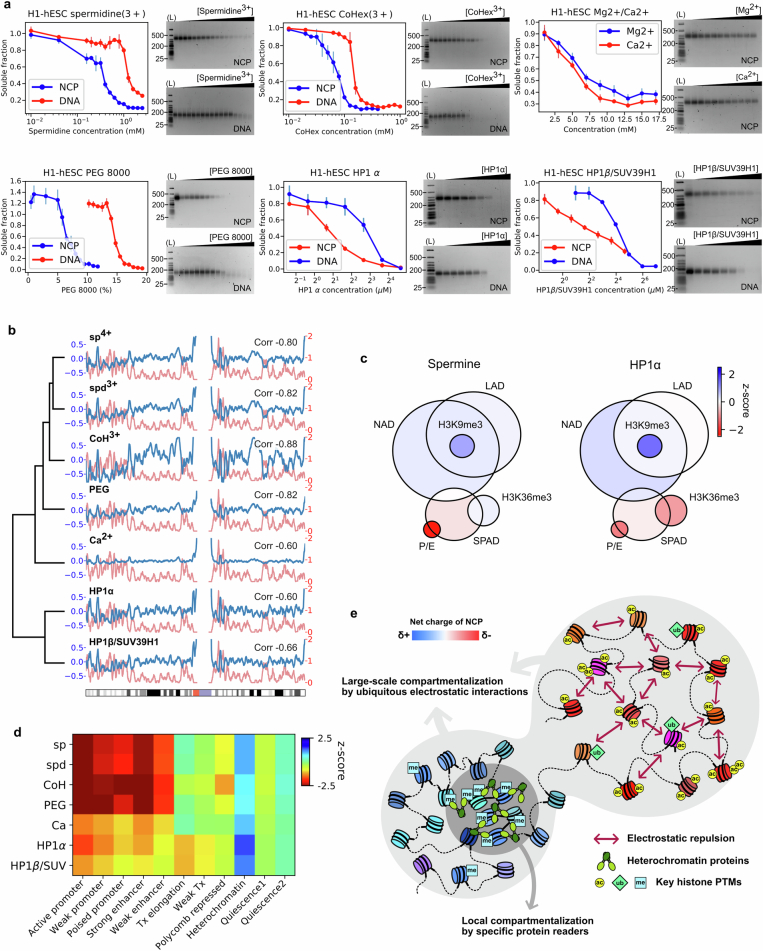
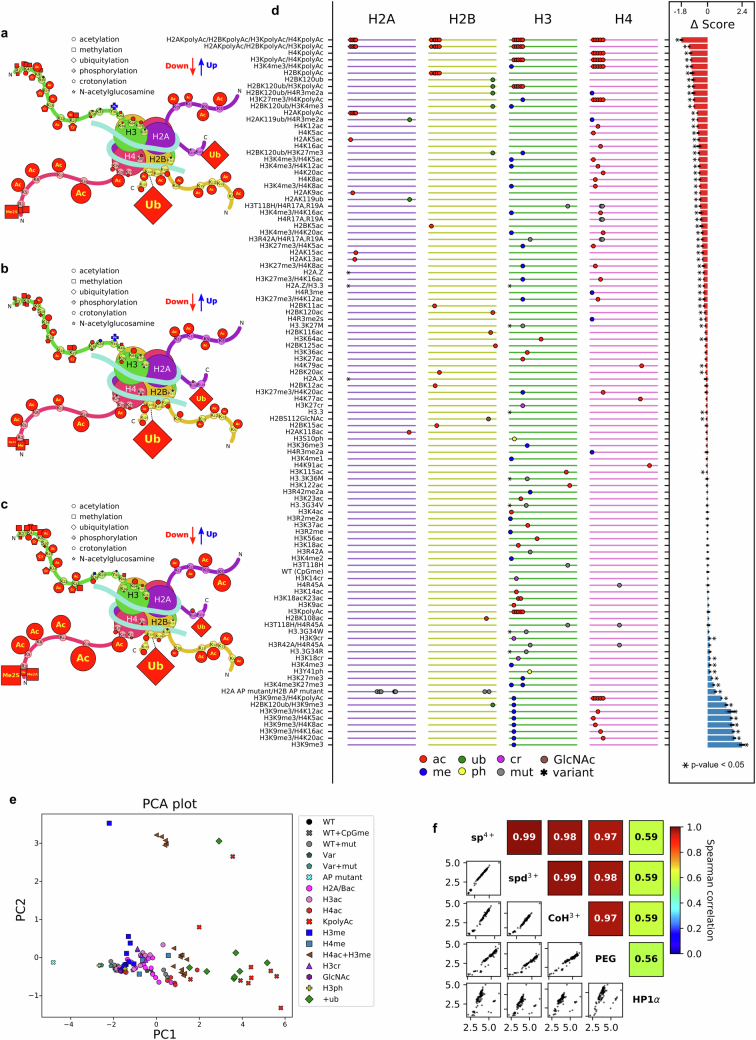
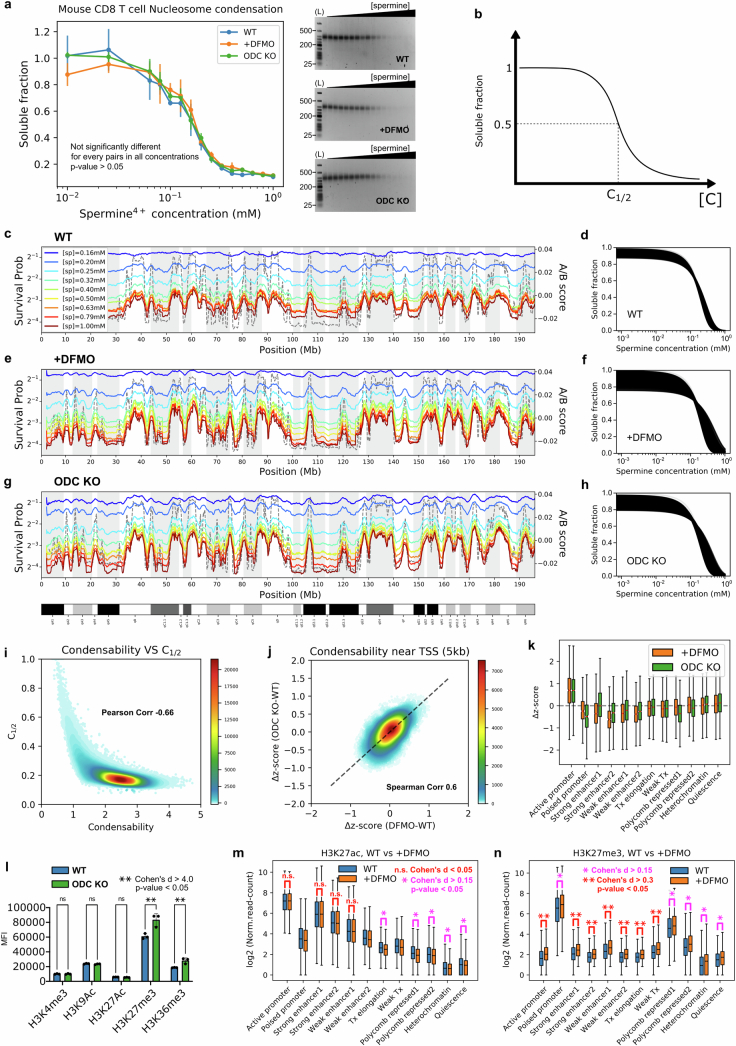
The eukaryotic genome is packed into nucleosomes of 147 base pairs around a histone core and is organized into euchromatin and heterochromatin, corresponding to the A and B compartments, respectively1,2. Here we investigated whether individual nucleosomes contain sufficient information for 3D genomic organization into compartments, for example, in their biophysical properties. We purified native mononucleosomes to high monodispersity and used physiological concentrations of polyamines to determine their condensability. The chromosomal regions known to partition into A compartments have low condensability and those for B compartments have high condensability. Chromatin polymer simulations using condensability as the only input, without any trans factors, reproduced the A/B compartments. Condensability is also strongly anticorrelated with gene expression, particularly near the promoters and in a cell type-dependent manner. Therefore, mononucleosomes have biophysical properties associated with genes being on or off. Comparisons with genetic and epigenetic features indicate that nucleosome condensability is an emergent property, providing a natural axis on which to project the high-dimensional cellular chromatin state. Analysis using various condensing agents or histone modifications and mutations indicates that the genome organization principle encoded into nucleosomes is mostly electrostatic in nature. Polyamine depletion in mouse T cells, resulting from either knocking out or inhibiting ornithine decarboxylase, results in hyperpolarized condensability, indicating that when cells cannot rely on polyamines to translate the biophysical properties of nucleosomes to 3D genome organization, they accentuate condensability contrast, which may explain the dysfunction observed with polyamine deficiency3-5.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Update of
-
Electrostatic encoding of genome organization principles within single native nucleosomes.bioRxiv [Preprint]. 2023 Dec 9:2023.12.08.570828. doi: 10.1101/2023.12.08.570828. bioRxiv. 2023. Update in: Nature. 2025 Jul;643(8071):572-581. doi: 10.1038/s41586-025-08971-7. PMID: 38106048 Free PMC article. Updated. Preprint.
Similar articles
-
Electrostatic encoding of genome organization principles within single native nucleosomes.bioRxiv [Preprint]. 2023 Dec 9:2023.12.08.570828. doi: 10.1101/2023.12.08.570828. bioRxiv. 2023. Update in: Nature. 2025 Jul;643(8071):572-581. doi: 10.1038/s41586-025-08971-7. PMID: 38106048 Free PMC article. Updated. Preprint.
-
Sexual Harassment and Prevention Training.2024 Mar 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Mar 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 36508513 Free Books & Documents.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
A rapid and systematic review of the clinical effectiveness and cost-effectiveness of paclitaxel, docetaxel, gemcitabine and vinorelbine in non-small-cell lung cancer.Health Technol Assess. 2001;5(32):1-195. doi: 10.3310/hta5320. Health Technol Assess. 2001. PMID: 12065068
Cited by
-
Polyamines buffer labile iron to suppress ferroptosis.bioRxiv [Preprint]. 2025 Jul 2:2025.06.30.662349. doi: 10.1101/2025.06.30.662349. bioRxiv. 2025. PMID: 40631118 Free PMC article. Preprint.
-
Non-disruptive 3D profiling of combinations of epigenetic marks in single cells.bioRxiv [Preprint]. 2025 Jun 17:2025.06.13.659535. doi: 10.1101/2025.06.13.659535. bioRxiv. 2025. PMID: 40666962 Free PMC article. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
