

Molecular mechanisms of programmed cell death and potential targeted pharmacotherapy in ischemic stroke (Review)
- PMID: 40341937
- PMCID: PMC12081036
- DOI: 10.3892/ijmm.2025.5544
Molecular mechanisms of programmed cell death and potential targeted pharmacotherapy in ischemic stroke (Review)
Abstract
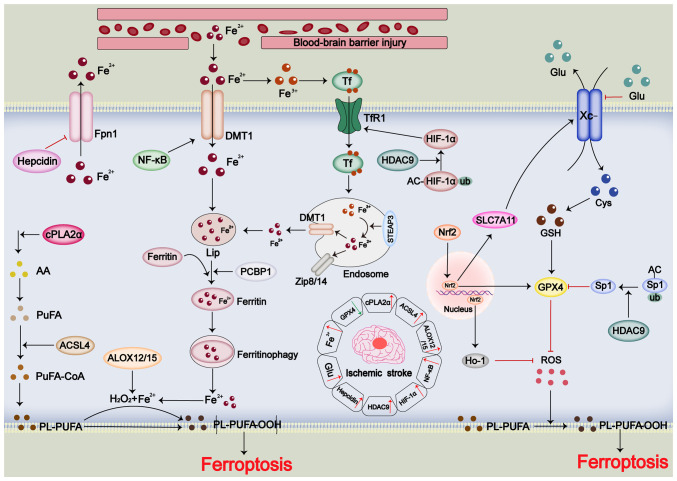
Stroke poses a threat to the elderly, being the second leading cause of death and the third leading cause of disability worldwide. Ischemic stroke (IS), resulting from arterial occlusion, accounts for ~85% of all strokes. The pathophysiological processes involved in IS are intricate and complex. Currently, tissue plasminogen activator (tPA) is the only Food and Drug Administration‑approved drug for the treatment of IS. However, due to its limited administration window and the risk of symptomatic hemorrhage, tPA is applicable to only ~10% of patients with stroke. Additionally, the reperfusion process associated with thrombolytic therapy can further exacerbate damage to brain tissue. Therefore, a thorough understanding of the molecular mechanisms underlying IS‑induced injury and the identification of potential protective agents is critical for effective IS treatment. Over the past few decades, advances have been made in exploring potential protective drugs for IS. The present review summarizes the specific mechanisms of various forms of programmed cell death (PCD) induced by IS and highlights potential protective drugs targeting different PCD pathways investigated over the last decade. The present review provides a theoretical foundation for basic research and insights for the development of pharmacotherapy for IS.
Keywords: IS; PCD; molecular mechanisms; pharmacotherapy; protective drugs.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Asparagine Endopeptidase Inhibition Attenuates Tissue Plasminogen Activator-Induced Brain Hemorrhagic Transformation After Ischemic Stroke.CNS Neurosci Ther. 2025 Mar;31(3):e70345. doi: 10.1111/cns.70345. CNS Neurosci Ther. 2025. PMID: 40116141 Free PMC article.
-
Phthalide derivative CD21 attenuates tissue plasminogen activator-induced hemorrhagic transformation in ischemic stroke by enhancing macrophage scavenger receptor 1-mediated DAMP (peroxiredoxin 1) clearance.J Neuroinflammation. 2021 Jun 24;18(1):143. doi: 10.1186/s12974-021-02170-7. J Neuroinflammation. 2021. PMID: 34162400 Free PMC article.
-
Targeted nano-delivery strategies for facilitating thrombolysis treatment in ischemic stroke.Drug Deliv. 2021 Dec;28(1):357-371. doi: 10.1080/10717544.2021.1879315. Drug Deliv. 2021. PMID: 33517820 Free PMC article.
-
Angiogenesis after ischemic stroke.Acta Pharmacol Sin. 2023 Jul;44(7):1305-1321. doi: 10.1038/s41401-023-01061-2. Epub 2023 Feb 24. Acta Pharmacol Sin. 2023. PMID: 36829053 Free PMC article. Review.
-
Advanced drug delivery system against ischemic stroke.J Control Release. 2022 Apr;344:173-201. doi: 10.1016/j.jconrel.2022.02.036. Epub 2022 Mar 4. J Control Release. 2022. PMID: 35248645 Review.
Cited by
-
Targeting Programmed Cell Death in Flap Ischemia/Reperfusion Injury.Biomolecules. 2025 Jun 20;15(7):911. doi: 10.3390/biom15070911. Biomolecules. 2025. PMID: 40723782 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous