

This is a preprint.
Multi-cohort cross-omics analysis reveals disease mechanisms and therapeutic targets in HTLV-1-associated myelopathy, a neglected retroviral neuroinflammatory disorder
- PMID: 40343334
- PMCID: PMC12060986
- DOI: 10.21203/rs.3.rs-5960764/v1
Multi-cohort cross-omics analysis reveals disease mechanisms and therapeutic targets in HTLV-1-associated myelopathy, a neglected retroviral neuroinflammatory disorder
Abstract
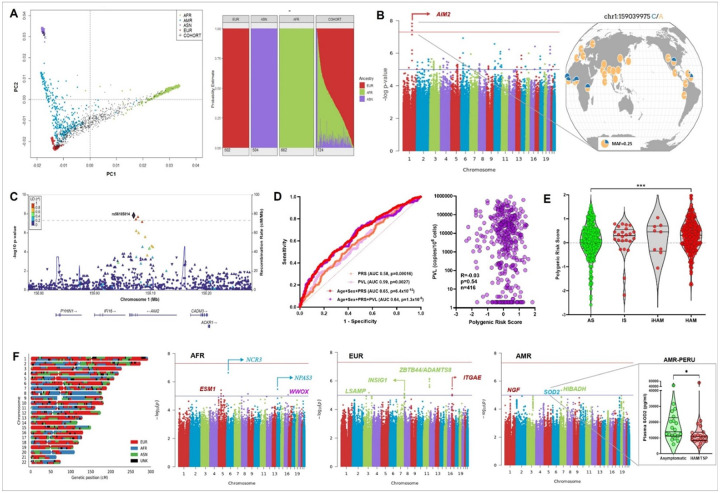
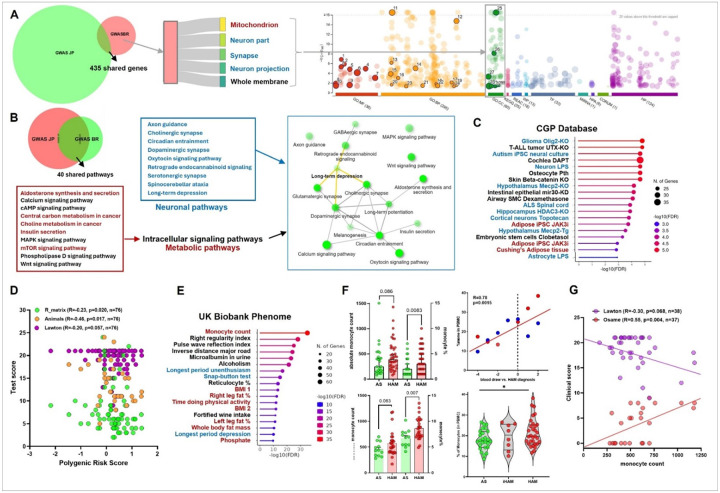
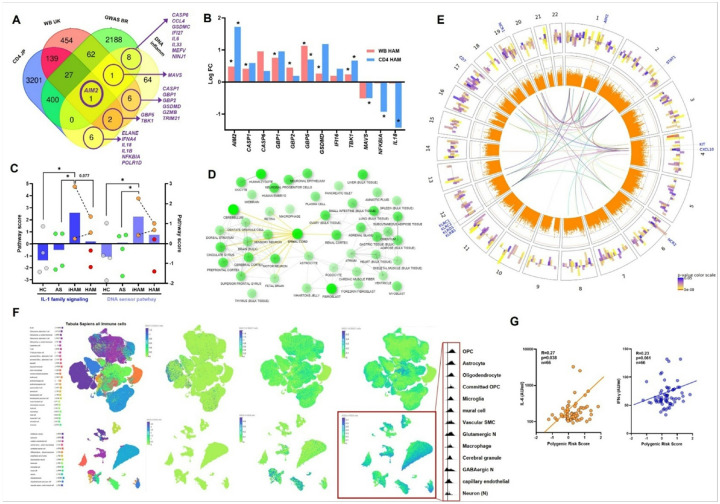
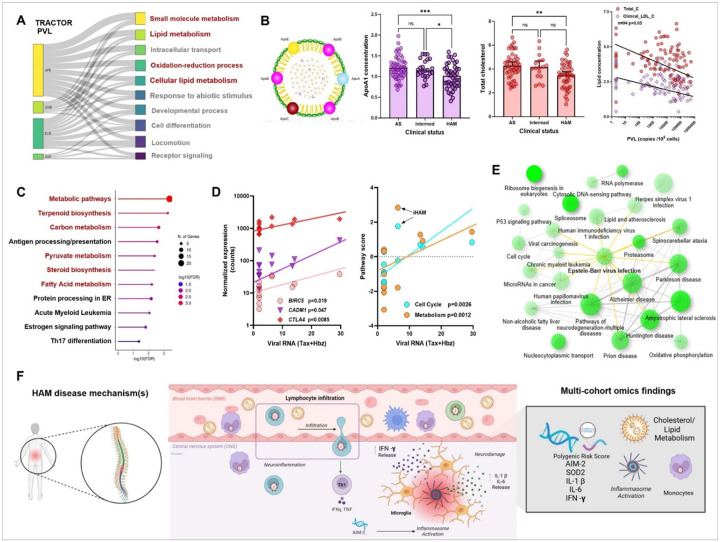
HTLV-1 is an enigmatic retrovirus triggering a debilitating neuroinflammatory disease, HTLV-1-associated myelopathy (HAM), with unknown pathogenesis. Both HTLV-1 infection and HAM predominantly affect women and non-white neglected populations. HAM is lacking disease-modifying treatment, as current treatment is mostly symptomatic and inspired by either HIV-1 or multiple sclerosis therapeutic strategies. We used systems biology analyses of novel and publicly available data comprising (epi)genomics, transcriptomics, metabolomics and proteomics of multi-ancestry cohorts from a total of > 2500 People Living with HTLV-1 from 5 countries (Brazil, Peru, Japan, UK, US). Leveraging an unique admixed Brazilian cohort, genome-wide association study (GWAS) revealed African-specific variants in inflammasome sensor AIM2 with genome-wide significance (p < 5x10-8). Suggestive loci (p > 5x10-8) corresponding to metabolic, immune and neuronal genes were validated using published Japanese GWAS. Polygenic risk score and proviral load were independent disease predictors across ancestries. Systems biology analysis revealed neuronal/synaptic signaling, monocyte count, glucose/lipid metabolism, and neurocognition/depression as genetically linked to HAM. In silico drug screening identified estrogen blocker Fulvestrant as the top hit, while also confirming existing (pre)clinical data for HDAC inhibitors and immunosuppressants. Validated GWAS genes were overexpressed in HAM patients' whole blood and CD4 T-cells, as well as in spinal cord astrocytes, oligodendrocytes, and microglia by single-cell RNAseq. We experimentally confirmed decreased ApoA1/lipid/cholesterol levels, higher monocyte levels and lower neurocognitive scores in multi-ancestry cohorts. We found striking biological similarities between retroviral Hbz/Tax overexpression, Hbz interactome and HAM multi-omics findings: enrichment for lipid/cholesterol metabolism, estrogen signaling, neurodegenerative diseases, and viral pathways including EBV, recently identified as the major driver of multiple sclerosis. In conclusion, our data-driven approach uncovers novel disease mechanisms and therapeutic targets, and a validated polygenic risk score allowing targeted surveillance for high-risk individuals. A strong molecular overlap to other neurodegenerative/neuroinflammatory diseases reveals shared neuropathogenic pathways between unrelated viruses.
Keywords: ancestry; multi-omics; neglected disease; neuroinflammation; retrovirus.
Conflict of interest statement
Conflict of Interests: All authors report no potential conflicts.
Figures
References
-
- Martin F., Tagaya Y. & Gallo R. Time to eradicate HTLV-1: an open letter to WHO. Lancet 391, 1893–1894 (2018). - PubMed
-
- Rosadas C. & Taylor G. P. Health inequities and HTLV-1. Lancet Microbe 3, e164 (2022). - PubMed
-
- Schierhout G. et al. Association between HTLV-1 infection and adverse health outcomes: a systematic review and meta-analysis of epidemiological studies. Lancet Infect Dis 20, 133–143 (2020). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
