

Integrating Pharmacokinetics and Quantitative Systems Pharmacology Approaches in Generative Drug Design
- PMID: 40343729
- PMCID: PMC12117666
- DOI: 10.1021/acs.jcim.5c00107
Integrating Pharmacokinetics and Quantitative Systems Pharmacology Approaches in Generative Drug Design
Abstract
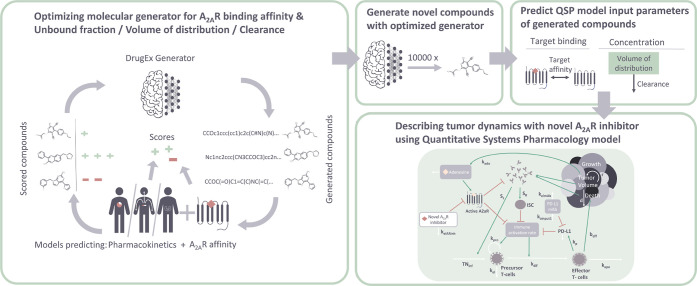
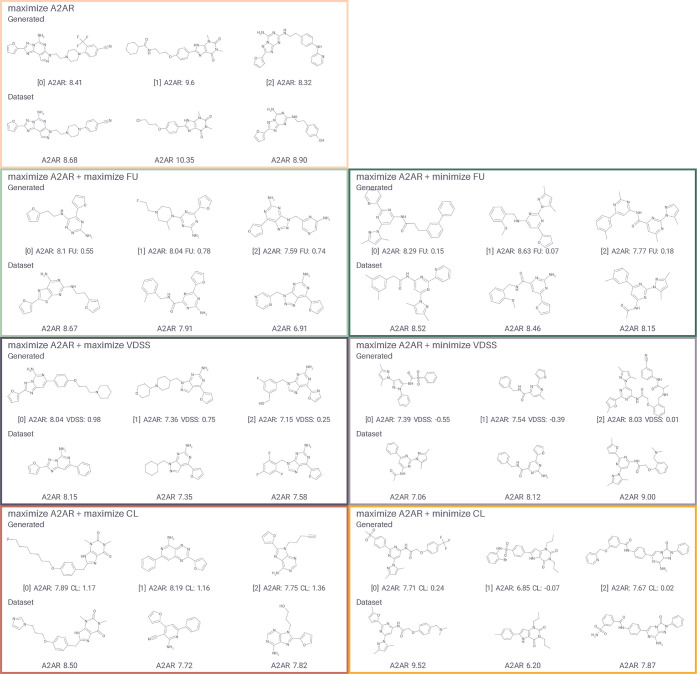
Integrated understanding of pharmacokinetics (PK) and pharmacodynamics (PD) is a key aspect of successful drug discovery. Yet in generative computational drug design, the focus often lies on optimizing potency. Here we integrate PK property predictions in DrugEx, a generative drug design framework and we explore the generated compounds' PD through simulations with a quantitative systems pharmacology (QSP) model. Quantitative structure-property relationship models were developed to predict molecule PK (clearance, volume of distribution and unbound fraction) and affinity for the Adenosine A2AR receptor (A2AR), a drug target in immuno-oncology. These models were used to score compounds in a reinforcement learning framework to generate molecules with a specific PK profile and high affinity for the A2AR. We predicted the expected tumor growth inhibition profiles using the QSP model for selected candidate molecules with varying PK and affinity profiles. We show that optimizing affinity to the A2AR, while minimizing or maximizing a PK property, shifts the type of molecular scaffolds that are generated. The difference in physicochemical properties of the compounds with different predicted PK parameters was found to correspond with the differences observed in the PK data set. We demonstrated the use of the QSP model by simulating the effect of a broad range of compound properties on the predicted tumor volume. In conclusion, our proposed integrated workflow incorporating affinity predictions with PKPD may provide a template for the next generation of advanced generative computational drug design.
Figures






References
-
- Chen E. P., Bondi R. W., Michalski P. J.. Model-based Target Pharmacology Assessment (mTPA): An Approach Using PBPK/PD Modeling and Machine Learning to Design Medicinal Chemistry and DMPK Strategies in Early Drug Discovery. J. Med. Chem. 2021;64:3185–3196. doi: 10.1021/acs.jmedchem.0c02033. - DOI - PubMed
-
- Gómez-Bombarelli R., Wei J. N., Duvenaud D., Hernández-Lobato J. M., Sánchez-Lengeling B., Sheberla D., Aguilera-Iparraguirre J., Hirzel T. D., Adams R. P., Aspuru-Guzik A.. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018;4:268–276. doi: 10.1021/acscentsci.7b00572. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
