

STIM2β is a Ca2+ signaling modulator for the regulation of mitotic clonal expansion and PPARG2 transcription in adipogenesis
- PMID: 40344531
- PMCID: PMC12326919
- DOI: 10.1111/febs.70118
STIM2β is a Ca2+ signaling modulator for the regulation of mitotic clonal expansion and PPARG2 transcription in adipogenesis
Abstract
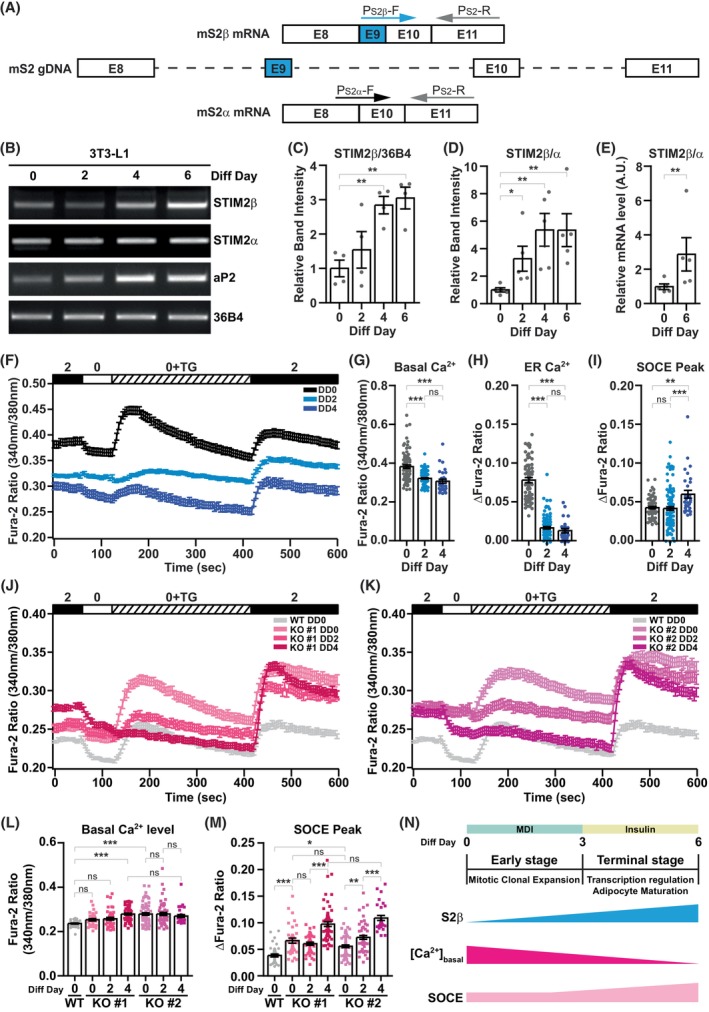
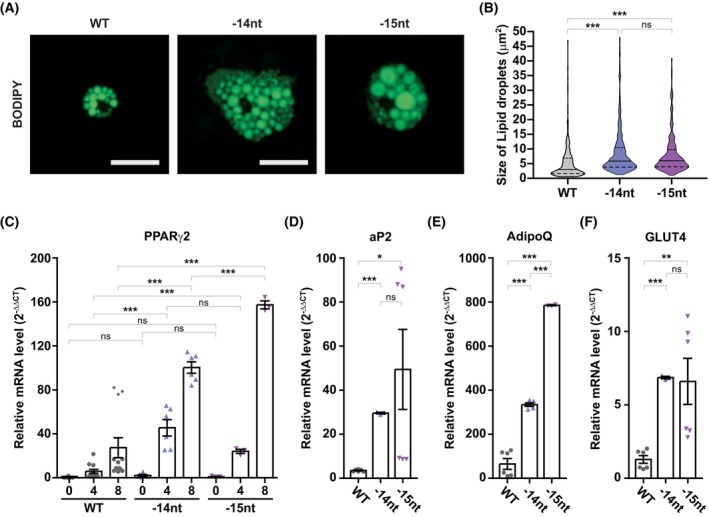
Intracellular Ca2+ is crucial in the regulation of adipocyte lipid metabolism and adipogenesis. In this study, we aimed to investigate the regulation mechanism of intracellular Ca2+ levels ([Ca2+]i) during adipocyte differentiation. We found that the expression of stromal interaction molecule 2 beta (STIM2β), which is the inhibitor of store-operated Ca2+ entry (SOCE), is upregulated throughout the differentiation process. Evaluation of [Ca2+]i in 3 T3-L1 and primary stromal vascular fraction (SVF) cells revealed that the basal Ca2+ level is downregulated after differentiation. Knockout (KO) of STIM2β in 3T3-L1 and primary SVF cells showed increased [Ca2+]i, indicating the involvement of STIM2β in the regulation of [Ca2+]i during adipogenesis. We further evaluated the function of STIM2β-mediated [Ca2+]i in early and terminal differentiation of adipogenesis. Analysis of cell proliferation rate during mitotic clonal expansion (MCE) in wild-type and STIM2β KO 3T3-L1 cell lines revealed that a larger population of KO cells underwent G1 arrest, suggesting that reduced [Ca2+]i by STIM2β induces MCE. Additionally, ablation of STIM2β increased differentiation efficiency, with more lipid accumulation and rapid transcriptional activation of adipogenic genes, especially proliferator-activator receptor γ2 (PPARG2). We found that PPARG2 transcription is regulated by store-operated calcium entry (SOCE) downstream transcription factors, confirming that increased [Ca2+]i by STIM2β ablation promotes PPARG2 transcription during adipogenesis. Additionally, STIM2β KO mice showed hypertrophic adipose tissue development. Our data suggest that STIM2β-mediated [Ca2+]i plays a pivotal role in the regulation of mitotic clonal expansion and PPARG2 gene activation and provides evidence that MCE is not a prerequisite process for terminal differentiation during adipogenesis.
Keywords: PPARγ2; STIM2β; adipogenesis; cell cycle regulation; intracellular Ca2+.
© 2025 The Author(s). The FEBS Journal published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Qiu Z, Wei Y, Chen N, Jiang M, Wu J & Liao K (2001) DNA synthesis and mitotic clonal expansion is not a required step for 3T3‐L1 Preadipocyte differentiation into adipocytes. J Biol Chem 276, 11988–11995. - PubMed
-
- Guo W, Zhang K‐M, Tu K, Li Y‐X, Zhu L, Xiao H‐S, Yang Y & Wu J‐R (2009) Adipogenesis licensing and execution are disparately linked to cell proliferation. Cell Res 19, 216–223. - PubMed
-
- Tang QQ & Lane MD (2012) Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem 81, 715–736. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
