

The multilayered transcriptional architecture of glioblastoma ecosystems
- PMID: 40346361
- PMCID: PMC12081307
- DOI: 10.1038/s41588-025-02167-5
The multilayered transcriptional architecture of glioblastoma ecosystems
Abstract
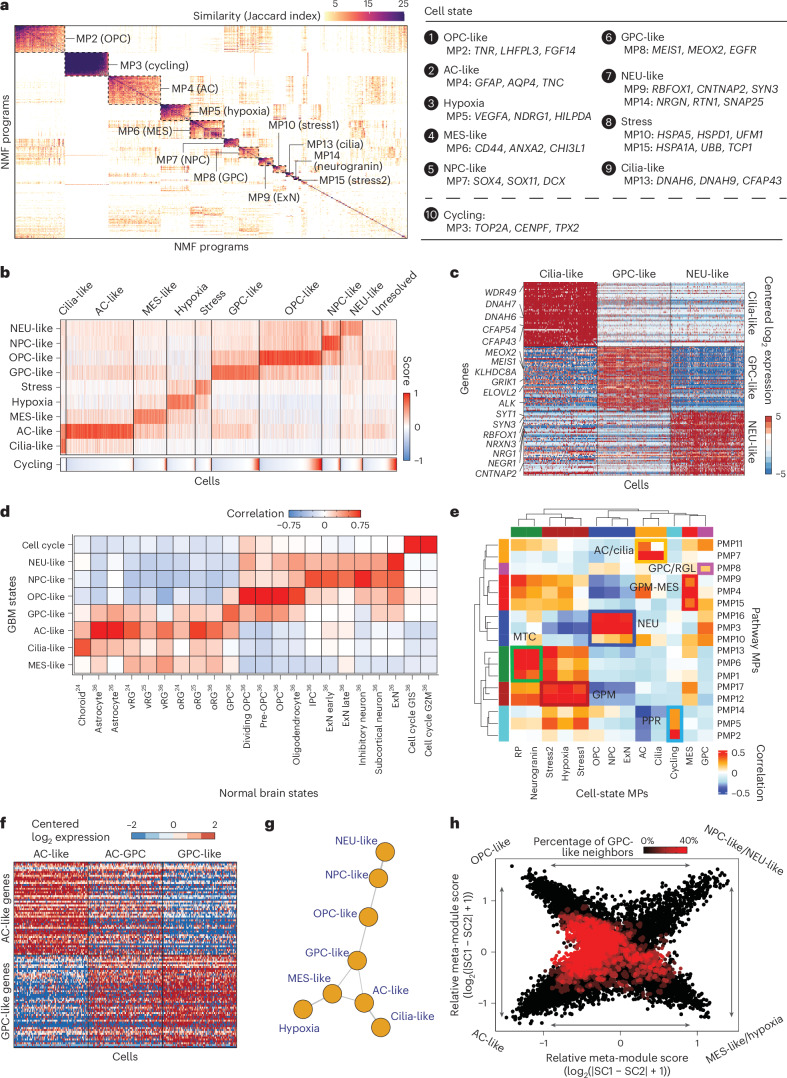
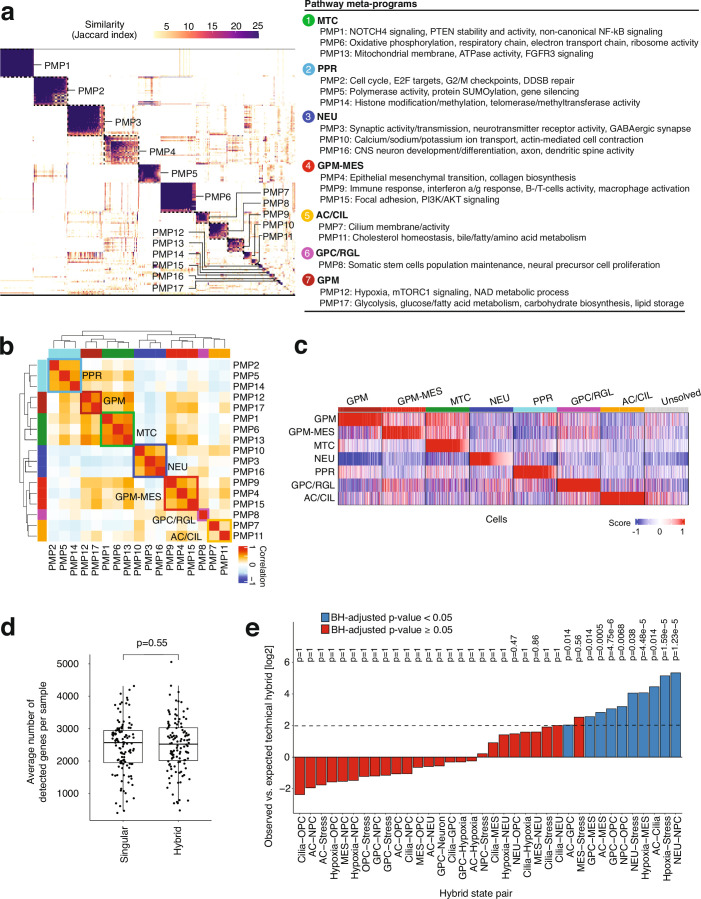
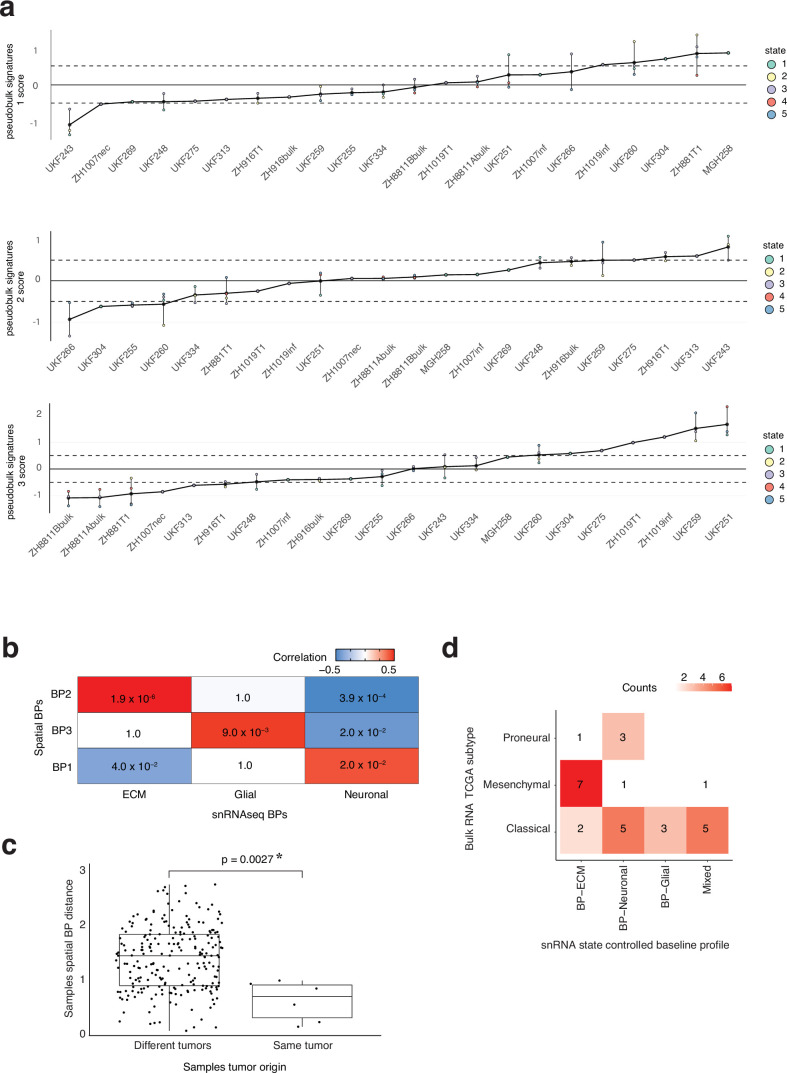
In isocitrate dehydrogenase wildtype glioblastoma (GBM), cellular heterogeneity across and within tumors may drive therapeutic resistance. Here we analyzed 121 primary and recurrent GBM samples from 59 patients using single-nucleus RNA sequencing and bulk tumor DNA sequencing to characterize GBM transcriptional heterogeneity. First, GBMs can be classified by their broad cellular composition, encompassing malignant and nonmalignant cell types. Second, in each cell type we describe the diversity of cellular states and their pathway activation, particularly an expanded set of malignant cell states, including glial progenitor cell-like, neuronal-like and cilia-like. Third, the remaining variation between GBMs highlights three baseline gene expression programs. These three layers of heterogeneity are interrelated and partially associated with specific genetic aberrations, thereby defining three stereotypic GBM ecosystems. This work provides an unparalleled view of the multilayered transcriptional architecture of GBM. How this architecture evolves during disease progression is addressed in the companion manuscript by Spitzer et al.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: M.L.S. is equity holder, scientific cofounder and advisory board member of Immunitas Therapeutics. I.T. is advisory board member of Immunitas Therapeutics and scientific cofounder of Cellyrix. R.G.W.V. is a cofounder and equity holder of Boundless Bio. The authors declare that such activities have no relationship to the present study. The other authors declare no competing interests.
Figures













References
-
- Venkataramani, V. et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell185, 2899–2917 (2022). - PubMed
MeSH terms
Substances
Grants and funding
- R37 CA245523/CA/NCI NIH HHS/United States
- R35 CA253183/CA/NCI NIH HHS/United States
- R01 CA239721/CA/NCI NIH HHS/United States
- R01 CA258763/CA/NCI NIH HHS/United States
- R01 CA237208/CA/NCI NIH HHS/United States
- P30 CA013696/CA/NCI NIH HHS/United States
- P30 CA240139/CA/NCI NIH HHS/United States
- P30 CA006516/CA/NCI NIH HHS/United States
- P50 CA165962/CA/NCI NIH HHS/United States
- R21 CA256575/CA/NCI NIH HHS/United States
- R21 NS114873/NS/NINDS NIH HHS/United States
- R01 CA276765/CA/NCI NIH HHS/United States
- R01 CA280560/CA/NCI NIH HHS/United States
- P30 CA034196/CA/NCI NIH HHS/United States
- R01 CA268592/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
