

Epidemiology of Gaucher Disease in France: Trends in Incidence, Mortality, Management, and Complications Over Three Decades
- PMID: 40348574
- PMCID: PMC12063198
- DOI: 10.1002/jimd.70037
Epidemiology of Gaucher Disease in France: Trends in Incidence, Mortality, Management, and Complications Over Three Decades
Abstract
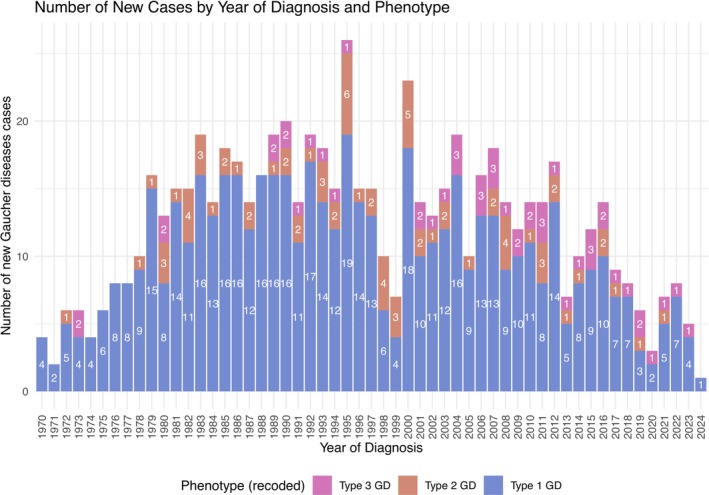
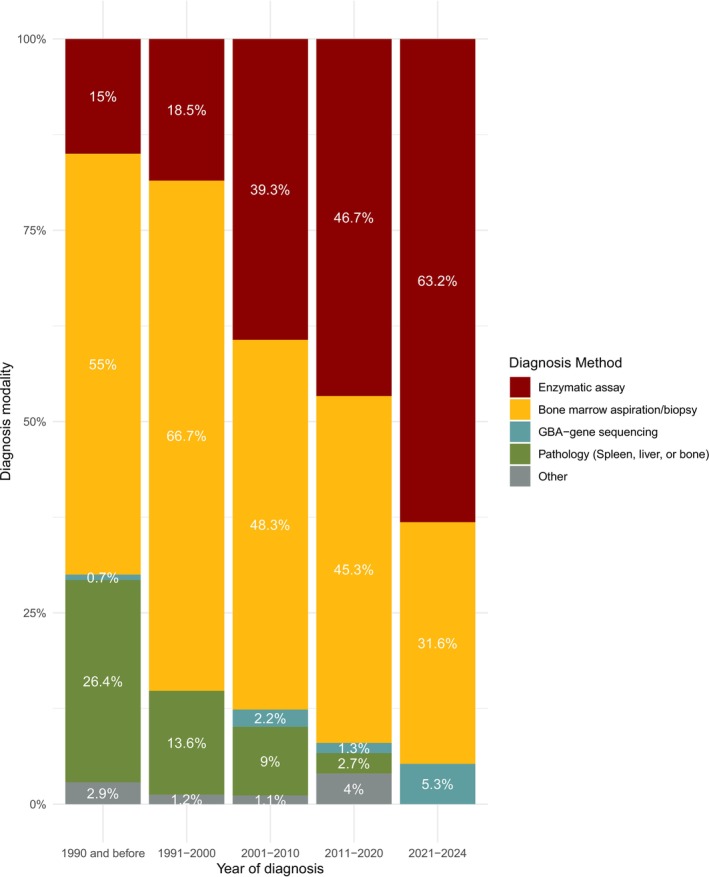
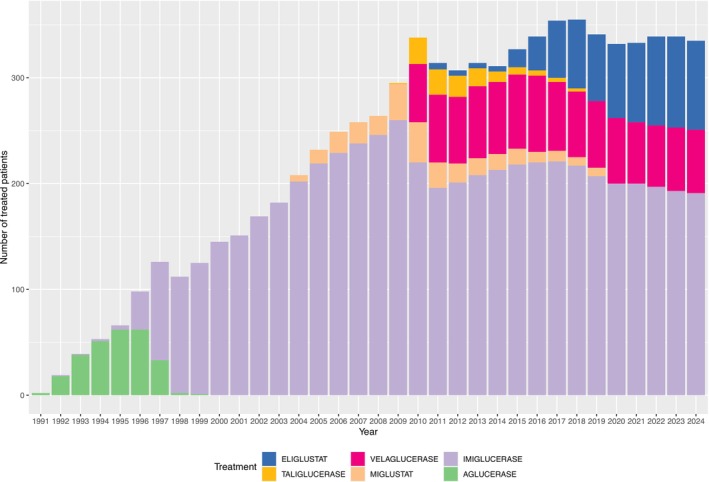
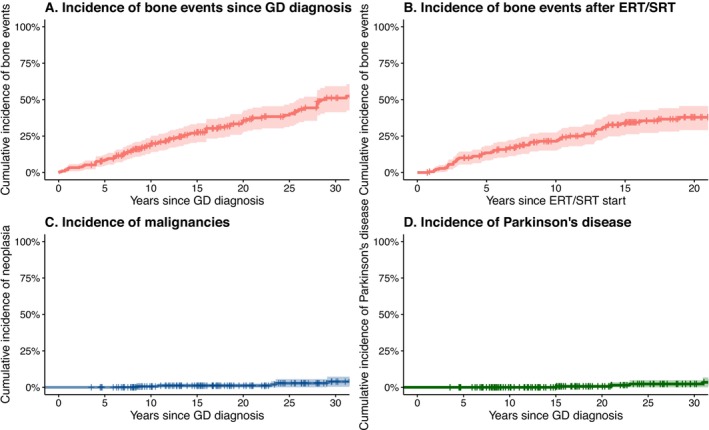
Gaucher disease (GD) is a rare autosomal-recessive lysosomal disorder caused by glucocerebrosidase deficiency. In this study, we described the epidemiology of GD in France over more than three decades. The French GD registry (FGDR) includes all known patients with GD in France. We described patients' characteristics, and estimated the incidence, prevalence, and standardized mortality ratios of GD. We compared the evolution of diagnostic methods, diagnosis delays, and treatment over time, and assessed the incidence of bone events, malignancies, and Parkinson's disease. Between 1980 and 2024, 706 confirmed GD were included. In 2024, 447 patients were alive (413 type 1, 34 type 3). GD incidence was 0.21/1 000 000 PY, and GD prevalence was 0.61 and 0.05/100 000 inhabitants for type 1 and 3, respectively. The standardized mortality ratio was 0.70 for type 1 GD and 16.23 for type 3 GD. Over time, we observed a decrease in the delay between first symptoms and diagnosis (5.4 years before 2000; 0.8 after 2020; p = 0.001), with enzyme assays becoming the primary diagnostic method, a reduction in splenectomies, and a gradual increase in the use of substrate reduction therapy in type 1 GD. The incidences of bone events, malignancies, and Parkinson's disease were 23, 2.7, and 1.07 per 1000 person-years, respectively. This study provides updated epidemiological data on GD in France, showing improvements in disease knowledge, faster and less invasive diagnoses, and reassuring outcomes for type 1 GD, with lower mortality and a relatively low incidence of malignancies and Parkinson's disease.
Keywords: Gaucher disease; enzyme replacement therapy; epidemiology; lysosomal disorder; registry; substrate reduction therapy.
© 2025 The Author(s). Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
Bénédicte Héron received travel fees from Sanofi. Anaïs Brassier received travel fees from Sanofi and Takeda, and consulting fees or other remuneration including fees as speaker from Sanofi and Takeda. Florence Dalbies received consulting fees or other remuneration, including a fee as a speaker from Sanofi. Bérengère Cador received travel fees from Sanofi and Takeda. Francis Gaches received travel fees from Sanofi. Agathe Masseau received a fee as a speaker from Sanofi and Takeda. Magali Pettazzoni received travel fees from Sanofi, and consulting fees or other remuneration including fees as speaker from Sanofi and Takeda. Wladimir Mauhin received fees for consultancy, speaking, travel grants, and meetings from Sanofi, Takeda, and Amicus therapeutics. Yann Nadjar received research grants from Takeda and received consulting fees or other remuneration, including fees as speaker, from Sanofi; assisting in the design of and/or participating in clinical studies using products manufactured by Sanofi. Christine Serratrice received consulting fees or other remuneration, including fees as a speaker from Takeda and Sanofi. Fabrice Camou received fees for consultancy, speaking, travel grants, and meetings from Sanofi and Takeda. Nadia Belmatoug received fees for consultancy, speaking, travel grants, and meetings from Sanofi and Takeda. The other authors declare no conflicts of interest.
Figures

References
-
- Brady R. O., Kanfer J. N., and Shapiro D., “Metabolism of Glucocerebrosides. II. Evidence of an Enzymatic Deficiency in Gaucher's Disease,” Biochemistry and Biophysics Research Communications 18 (1965): 221–225. - PubMed
-
- Nalysnyk L., Rotella P., Simeone J. C., Hamed A., and Weinreb N., “Gaucher Disease Epidemiology and Natural History: A Comprehensive Review of the Literature,” Hematology Amsterdam 22, no. 2 (2017): 65–73. - PubMed
-
- Nguyen Y., Stirnemann J., and Belmatoug N., “La maladie de Gaucher: quand y penser?,” Rev Médecine Interne 40 (2019): 313–322. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
