

Designer artificial environments for membrane protein synthesis
- PMID: 40348791
- PMCID: PMC12065789
- DOI: 10.1038/s41467-025-59471-1
Designer artificial environments for membrane protein synthesis
Abstract
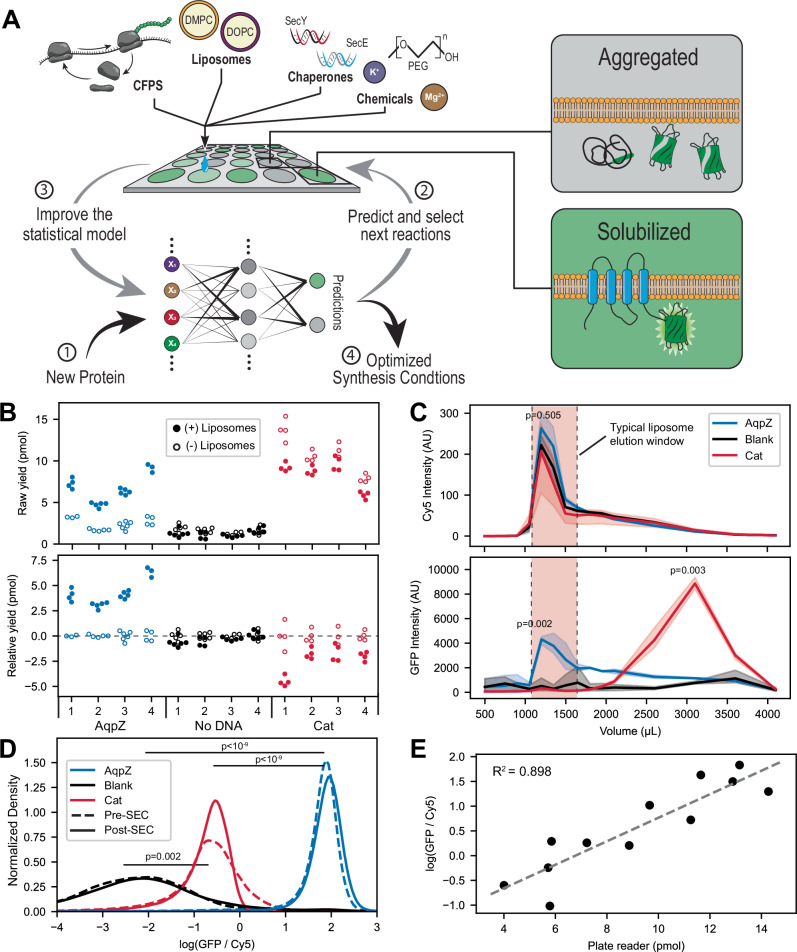
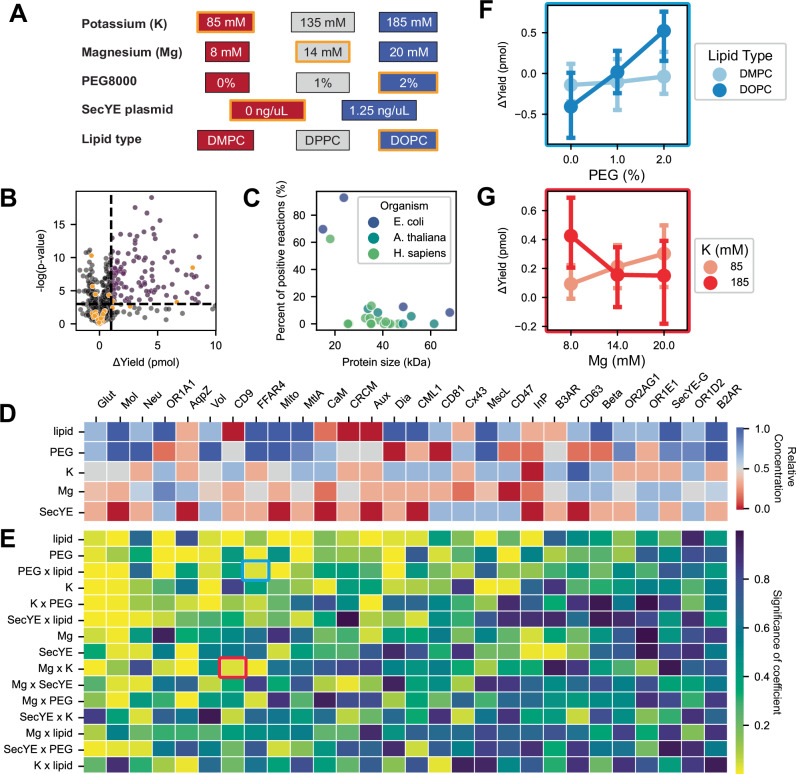
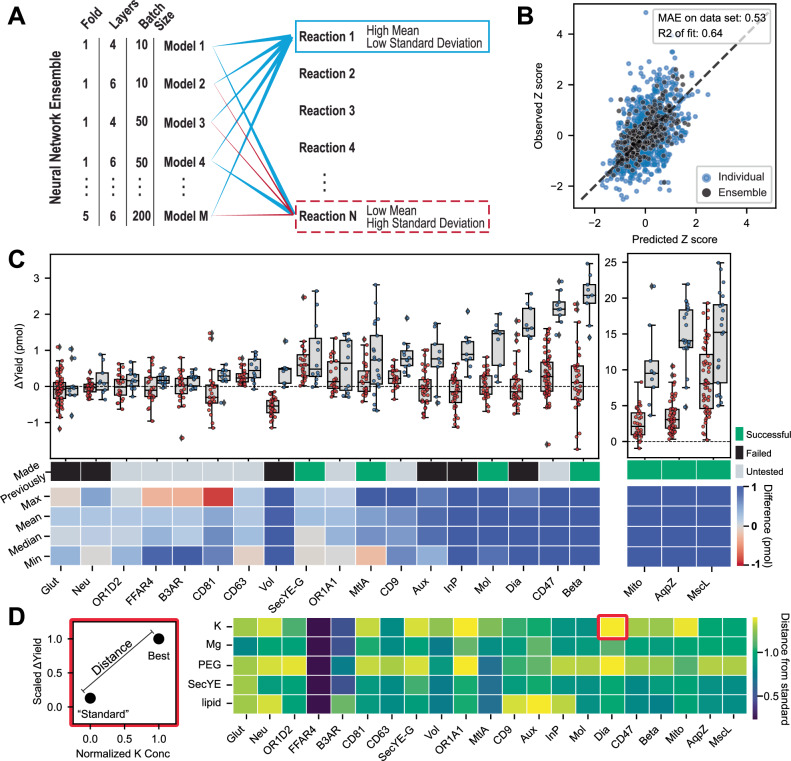
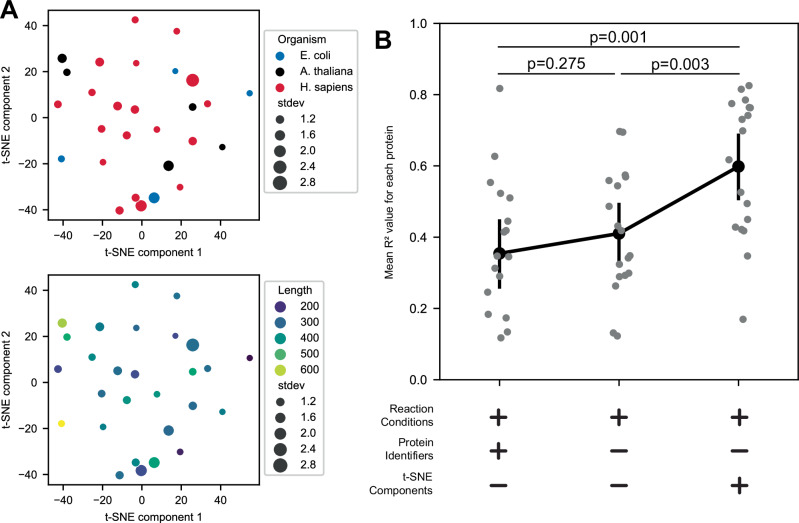
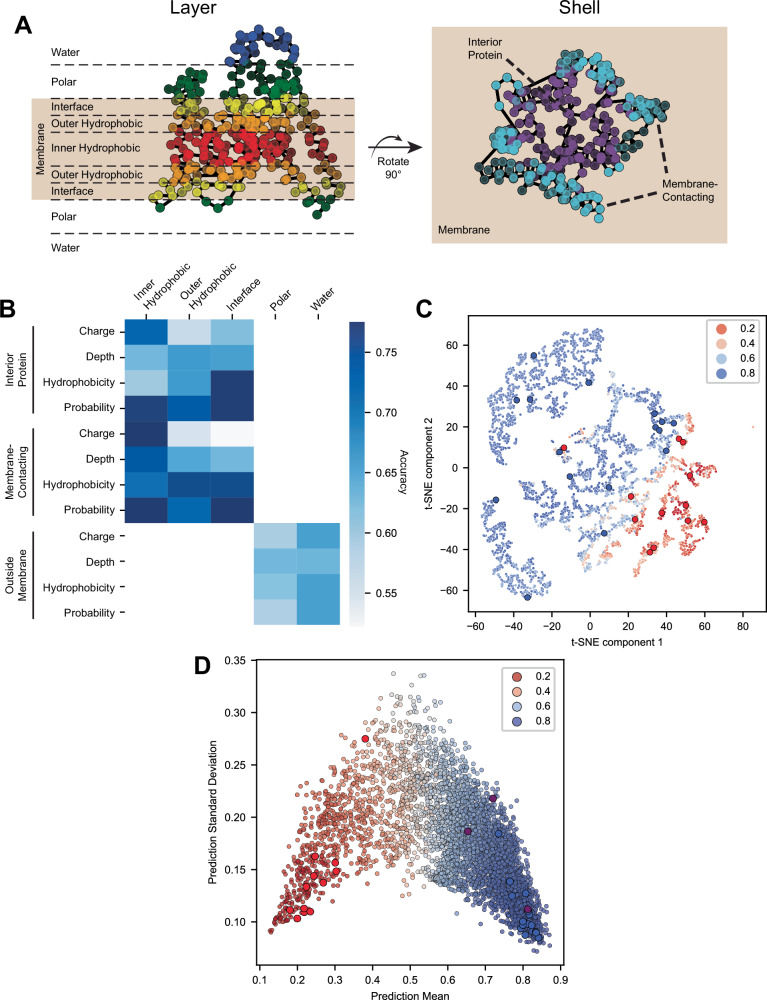
Protein synthesis in natural cells involves intricate interactions between chemical environments, protein-protein interactions, and protein machinery. Replicating such interactions in artificial and cell-free environments can control the precision of protein synthesis, elucidate complex cellular mechanisms, create synthetic cells, and discover new therapeutics. Yet, creating artificial synthesis environments, particularly for membrane proteins, is challenging due to the poorly defined chemical-protein-lipid interactions. Here, we introduce MEMPLEX (Membrane Protein Learning and Expression), which utilizes machine learning and a fluorescent reporter to rapidly design artificial synthesis environments of membrane proteins. MEMPLEX generates over 20,000 different artificial chemical-protein environments spanning 28 membrane proteins. It captures the interdependent impact of lipid types, chemical environments, chaperone proteins, and protein structures on membrane protein synthesis. As a result, MEMPLEX creates new artificial environments that successfully synthesize membrane proteins of broad interest but previously intractable. In addition, we identify a quantitative metric, based on the hydrophobicity of the membrane-contacting amino acids, that predicts membrane protein synthesis in artificial environments. Our work allows others to rapidly study and resolve the "dark" proteome using predictive generation of artificial chemical-protein environments. Furthermore, the results represent a new frontier in artificial intelligence-guided approaches to creating synthetic environments for protein synthesis.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- R01 EB034279/EB/NIBIB NIH HHS/United States
- R35 GM142788/GM/NIGMS NIH HHS/United States
- R35GM142788/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- 5R01EB034279/U.S. Department of Health & Human Services | NIH | National Institute of Biomedical Imaging and Bioengineering (NIBIB)
LinkOut - more resources
Full Text Sources
Research Materials
