

Recombinant Protein Spectral Library (rPSL) DIA-MS method improves identification and quantification of low-abundance cancer-associated and kynurenine pathway proteins
- PMID: 40348885
- PMCID: PMC12065878
- DOI: 10.1038/s42004-025-01531-0
Recombinant Protein Spectral Library (rPSL) DIA-MS method improves identification and quantification of low-abundance cancer-associated and kynurenine pathway proteins
Abstract
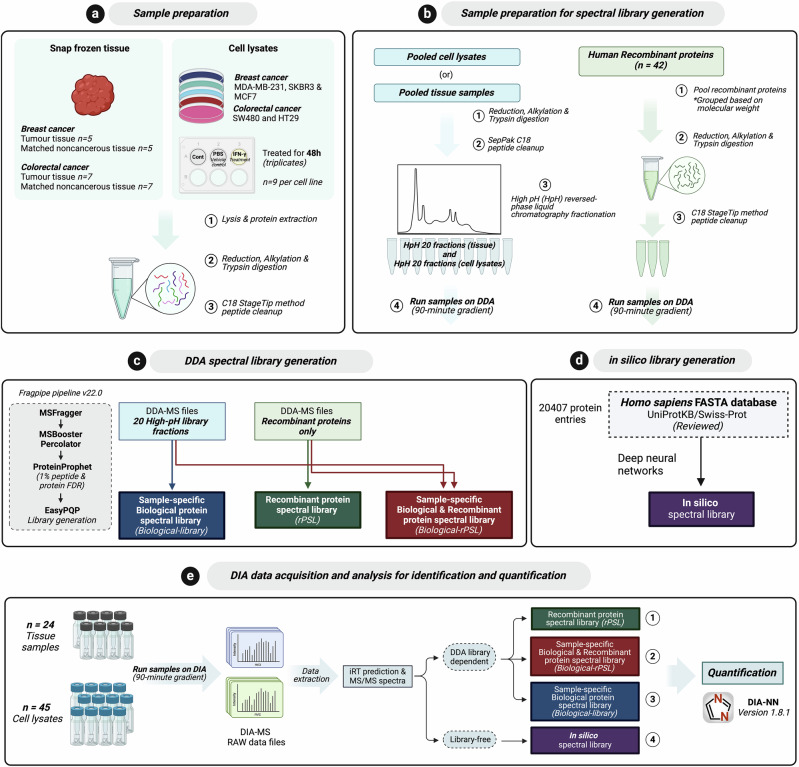
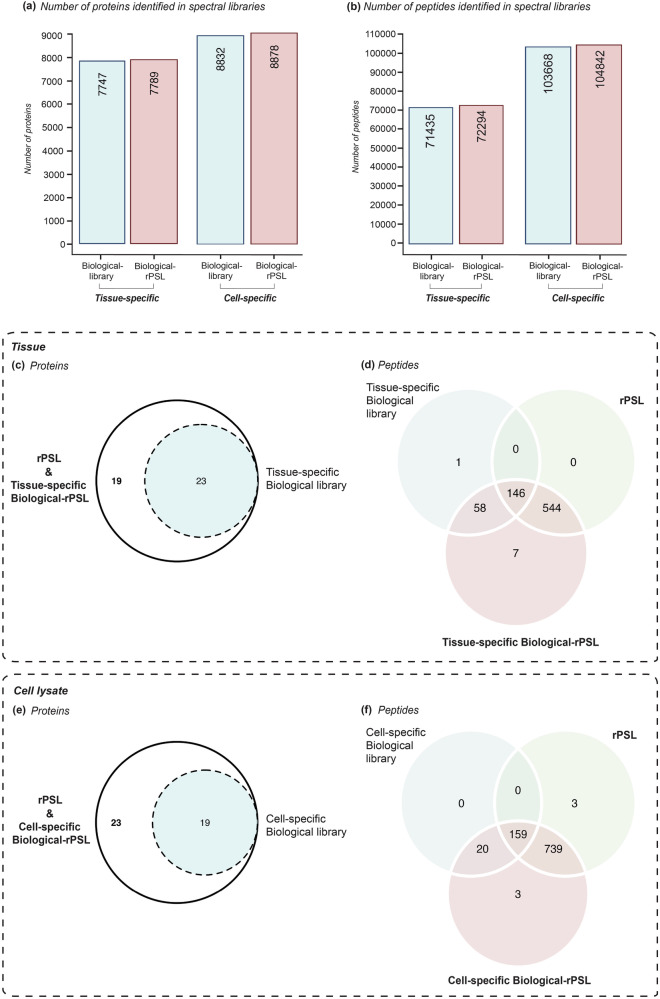
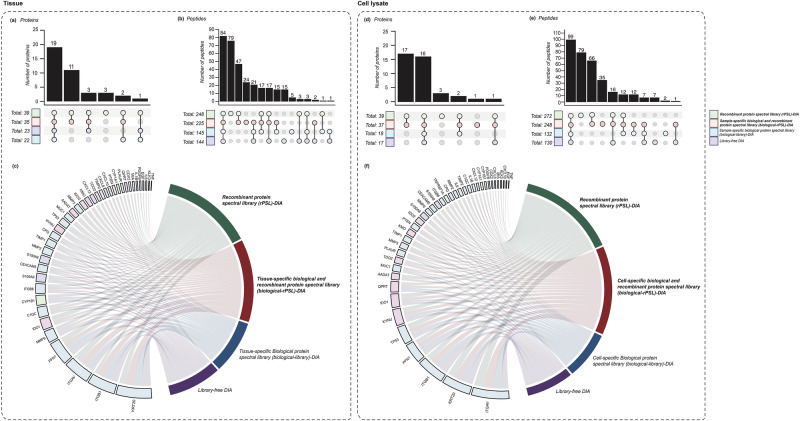
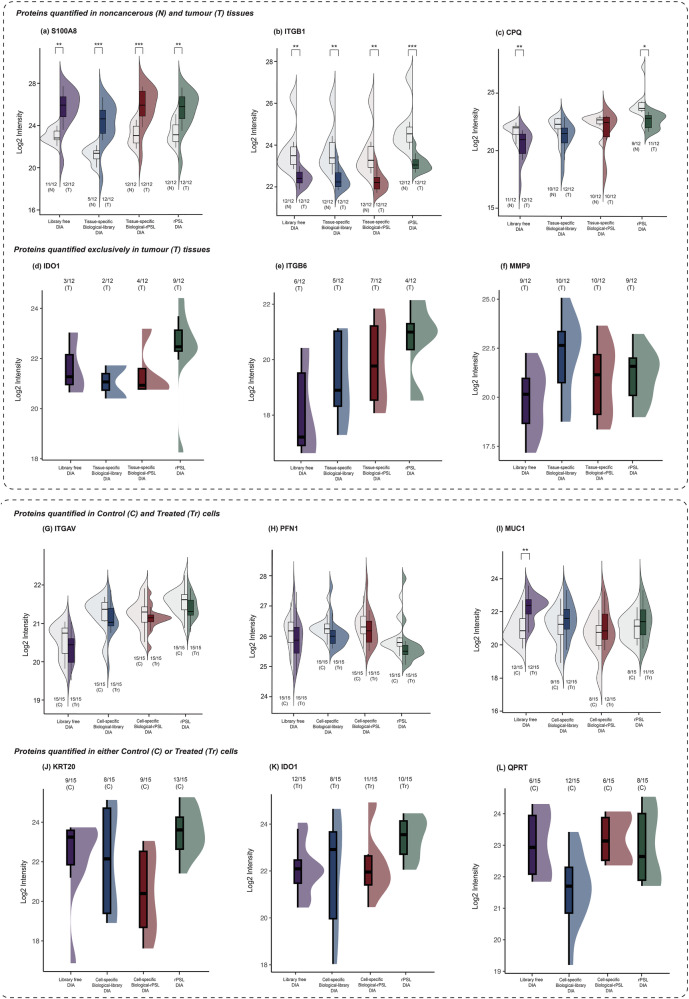
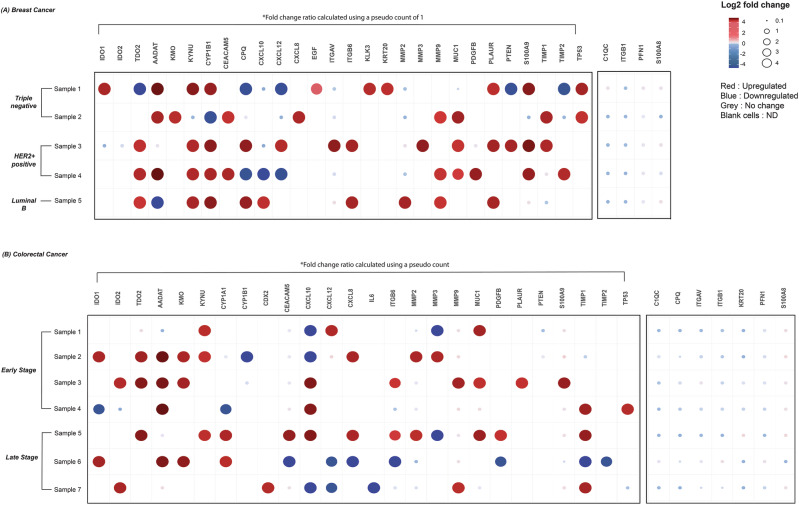
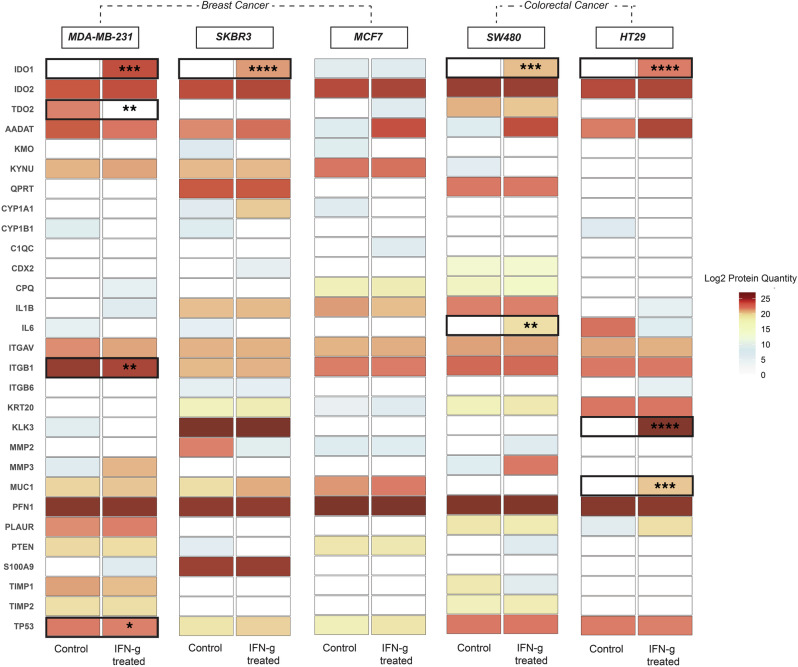
Data-independent acquisition mass spectrometry (DIA-MS) is a powerful tool for quantitative proteomics, but a well-constructed reference spectral library is crucial to optimize DIA analysis, particularly for low-abundance proteins. In this study, we evaluate the efficacy of a recombinant protein spectral library (rPSL), generated from tryptic digestion of 42 human recombinant proteins, in enhancing the detection and quantification of lower-abundance cancer-associated proteins. Additionally, we generated a combined sample-specific biological-rPSL by integrating the rPSL with a spectral library derived from pooled biological samples. We compared the performance of these libraries for DIA data extraction with standard methods, including sample-specific biological spectral library and library-free DIA methods. Our specific focus was on quantifying cancer-associated proteins, including key enzymes involved in kynurenine pathway, across patient-derived tissues and cell lines. Both rPSL and biological-rPSL-DIA approaches provided significantly improved coverage of lower-abundance proteins, enhancing sensitivity and more consistent protein quantification across matched tumour and adjacent noncancerous tissues from breast and colorectal cancer patients and in cancer cell lines. Overall, our study demonstrates that rPSL and biological-rPSL coupled with DIA-MS workflows, can address the limitations of both biological library-based and library-free DIA methods, offering a robust approach for quantifying low-abundance cancer-associated proteins in complex biological samples.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
