

Temporally discordant chromatin accessibility and DNA demethylation define short- and long-term enhancer regulation during cell fate specification
- PMID: 40349339
- PMCID: PMC12153380
- DOI: 10.1016/j.celrep.2025.115680
Temporally discordant chromatin accessibility and DNA demethylation define short- and long-term enhancer regulation during cell fate specification
Abstract
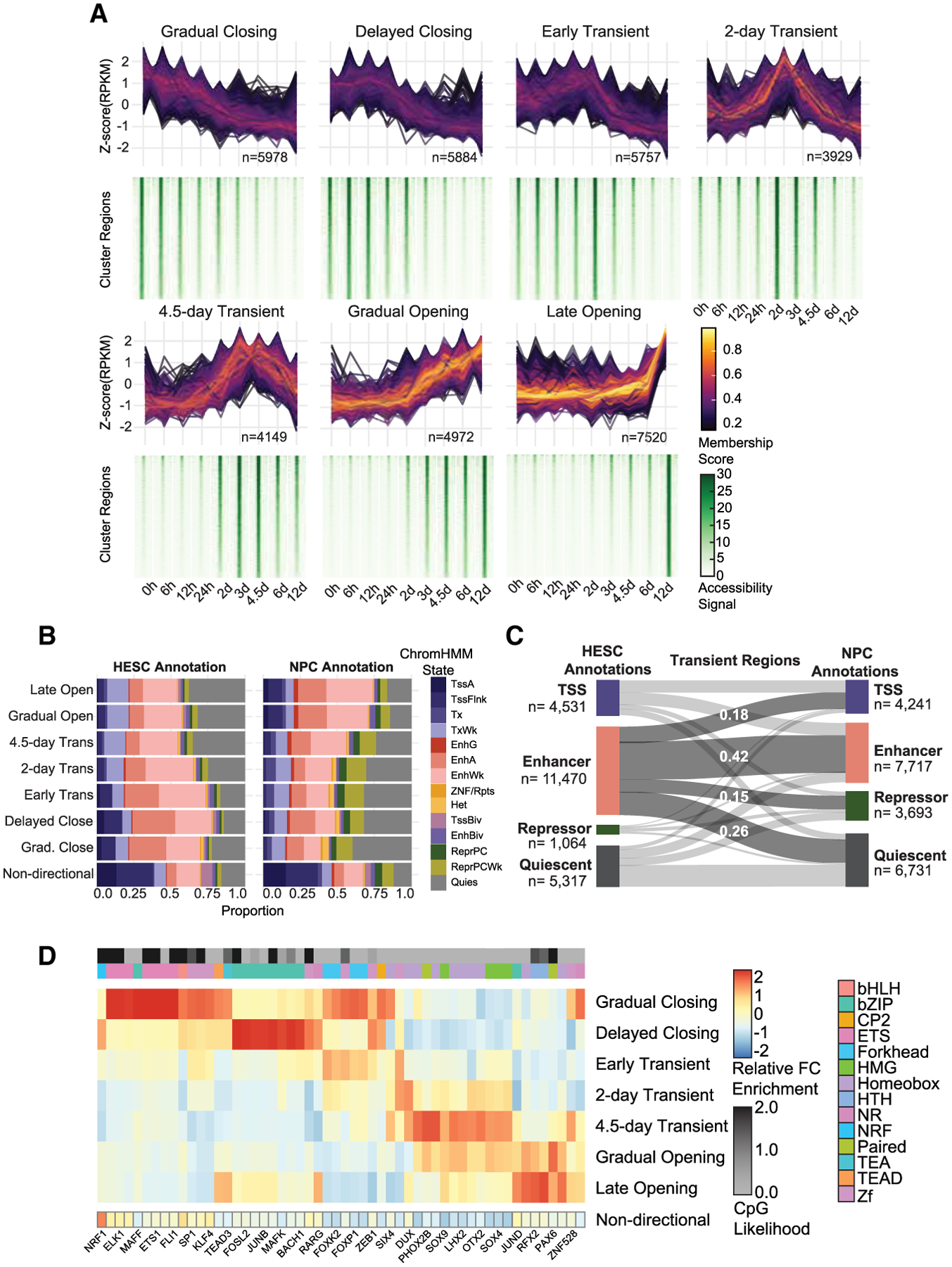
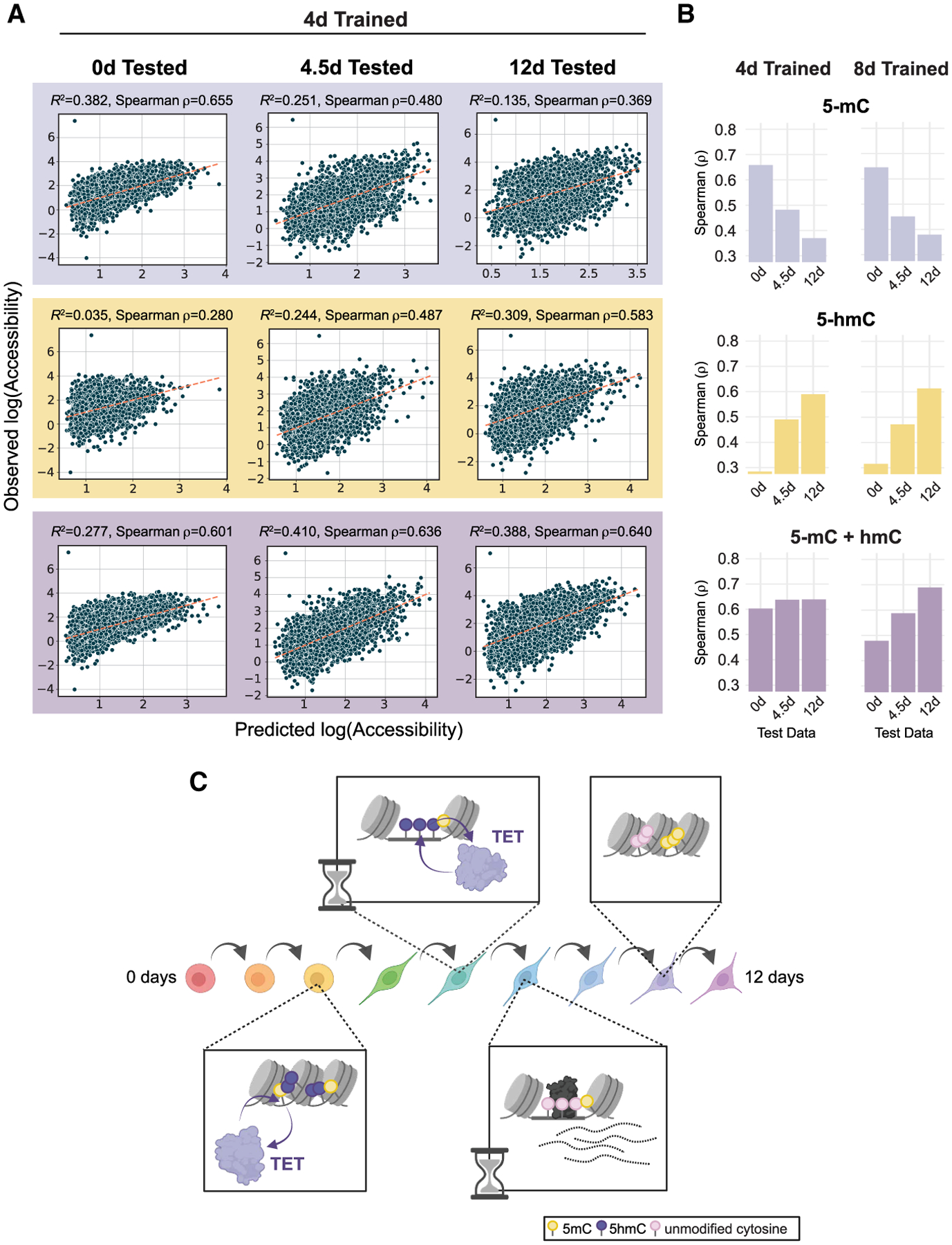
Chromatin and DNA modifications mediate the transcriptional activity of lineage-specifying enhancers, but recent work challenges the dogma that joint chromatin accessibility and DNA demethylation are prerequisites for transcription. To understand this paradox, we established a highly resolved timeline of their dynamics during neural progenitor cell differentiation. We discovered that, while complete demethylation appears delayed relative to shorter-lived chromatin changes for thousands of enhancers, DNA demethylation actually initiates with 5-hydroxymethylation before appreciable accessibility and transcription factor occupancy is observed. The extended timeline of DNA demethylation creates temporal discordance appearing as heterogeneity in enhancer regulatory states. Few regions ever gain methylation, and resulting enhancer hypomethylation persists long after chromatin activities have dissipated. We demonstrate that the temporal methylation status of CpGs (mC/hmC/C) predicts past, present, and future chromatin accessibility using machine learning models. Thus, chromatin and DNA methylation collaborate on different timescales to shape short- and long-term enhancer regulation during cell fate specification.
Keywords: 5-hydroxymethylation; 6-base sequencing; ATAC-Me; CP: Developmental biology; CP: Molecular biology; DNA methylation; chromatin accessibility; differentiation; enhancers; epigenetics; machine learning; neural progenitor cells.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests F.P., A.J., and T.C. are employees of biomodal, formerly Cambridge Epigenetix.
Figures




Update of
-
Temporally discordant chromatin accessibility and DNA demethylation define short and long-term enhancer regulation during cell fate specification.bioRxiv [Preprint]. 2024 Aug 27:2024.08.27.609789. doi: 10.1101/2024.08.27.609789. bioRxiv. 2024. Update in: Cell Rep. 2025 May 27;44(5):115680. doi: 10.1016/j.celrep.2025.115680. PMID: 39253426 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
