

Basal-shift transformation leads to EGFR therapy-resistance in human lung adenocarcinoma
- PMID: 40350470
- PMCID: PMC12066730
- DOI: 10.1038/s41467-025-59623-3
Basal-shift transformation leads to EGFR therapy-resistance in human lung adenocarcinoma
Abstract
Although EGFR tyrosine kinase inhibitors (EGFR-TKIs) are effective for EGFR-mutant lung adenocarcinoma (LUAD), resistance inevitably develops through diverse mechanisms, including secondary genetic mutations, amplifications and as-yet undefined processes. To comprehensively unravel the mechanisms of EGFR-TKI resistance, we establish a biobank of patient-derived EGFR-mutant lung cancer organoids, encompassing cases previously treated with EGFR-TKIs. Through comprehensive molecular profiling including single-cell analysis, here we identify a subgroup of EGFR-TKI-resistant LUAD organoids that lacks known resistance-related genetic lesions and instead exhibits a basal-shift phenotype characterized by the hybrid expression of LUAD- and squamous cell carcinoma-related genes. Prospective gene engineering demonstrates that NKX2-1 knockout induces the basal-shift transformation along with EGFR-target therapy resistance. Basal-shift LUADs frequently harbor CDKN2A/B loss and are sensitive to CDK4/6 inhibitors. Our EGFR-mutant lung cancer organoid library not only offers a valuable resource for lung cancer research but also provides insights into molecular underpinnings of EGFR-TKI resistance, facilitating the development of therapeutic strategies.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: T.S. is an inventor on several patents related to organoid culture. We declare that none of the authors have competing financial or non-financial interests as defined by Nature Portfolio.
Figures
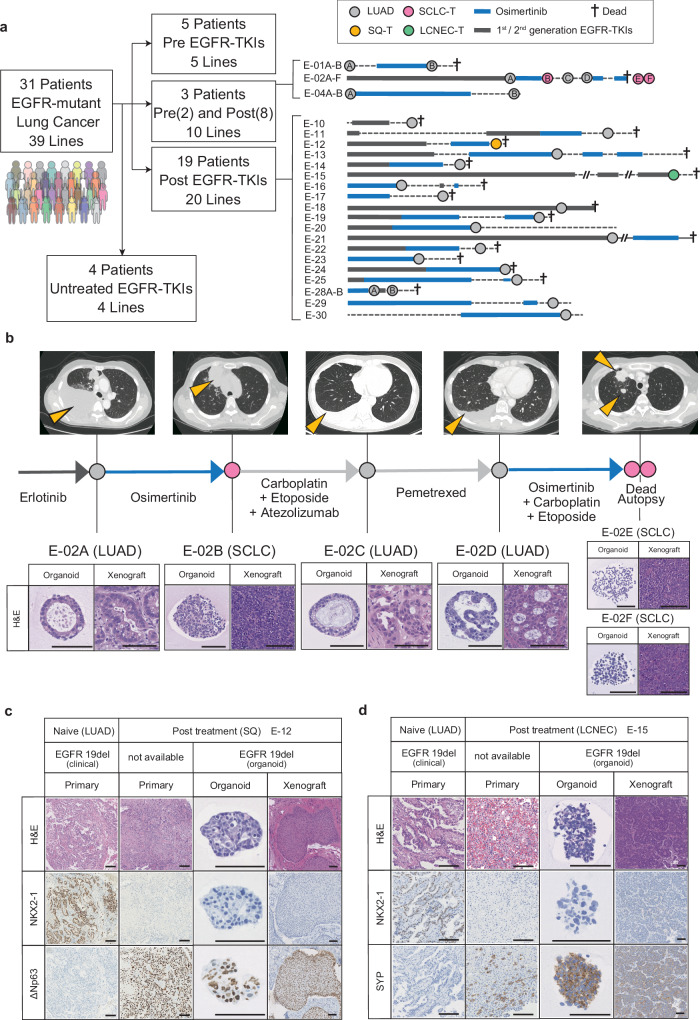
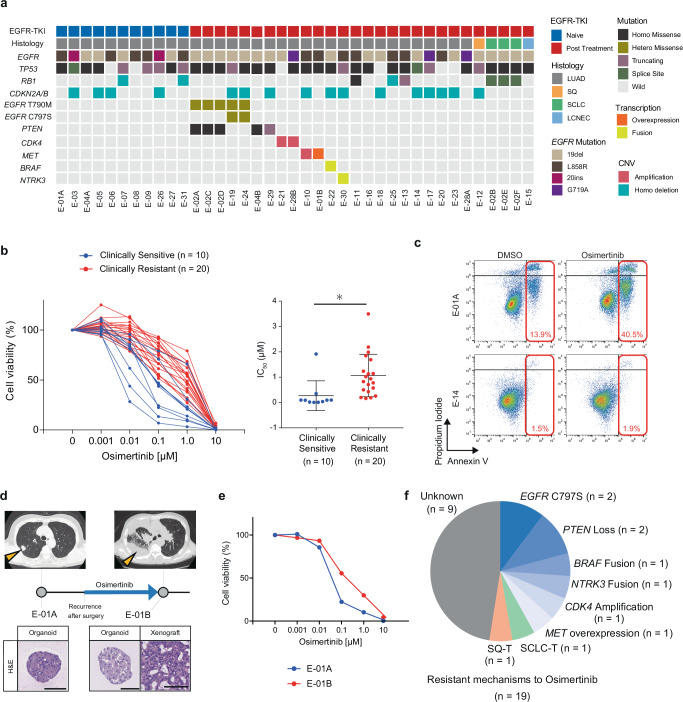
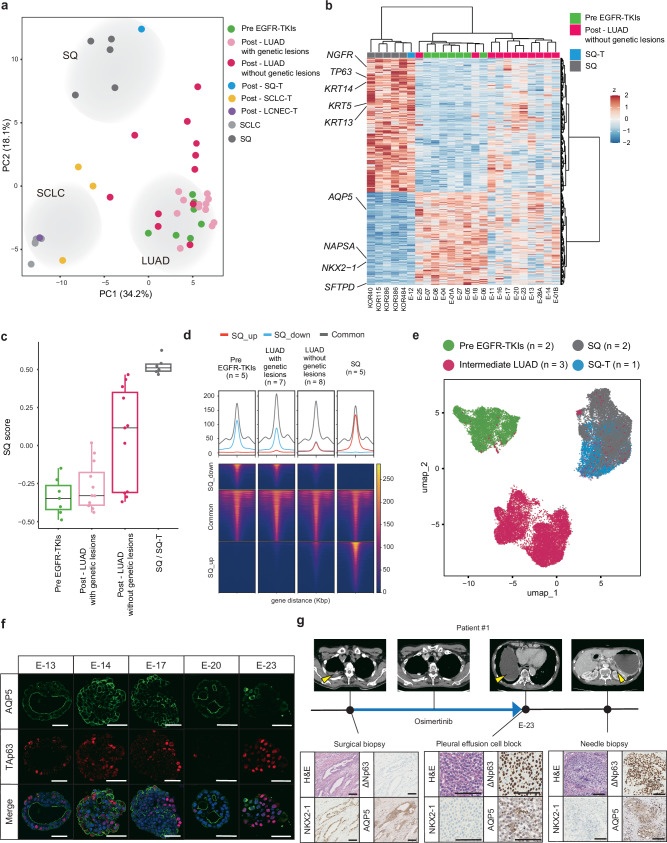

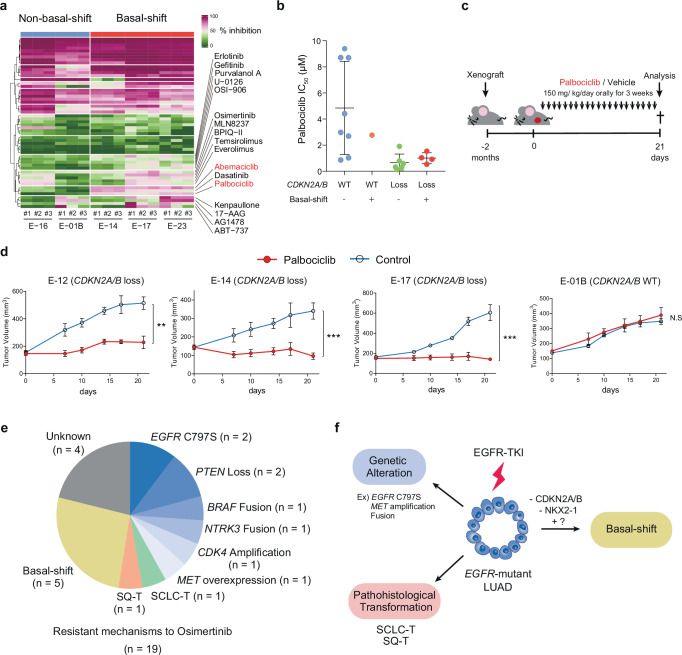
References
-
- Paez, J. G. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science304, 1497–1500 (2004). - PubMed
-
- Lynch, T. J. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med.350, 2129–2139 (2004). - PubMed
-
- Mok, T. S. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med.361, 947–957 (2009). - PubMed
-
- Rosell, R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol.13, 239–246 (2012). - PubMed
-
- Sequist, L. V. et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol.31, 3327–3334 (2013). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
